O variational quantum eigensolver (VQE)
Esta lição vai apresentar o variational quantum eigensolver, explicar sua importância como um algoritmo fundamental na computação quântica e também explorar seus pontos fortes e fracos. O VQE por si só, sem métodos de auxílio, provavelmente não é suficiente para computações quânticas modernas em escala de utilidade. Mesmo assim, ele é importante como um método híbrido clássico-quântico arquetípico, e serve de base para muitos algoritmos mais avançados.
Este vídeo traz uma visão geral do VQE e dos fatores que afetam sua eficiência. O texto abaixo adiciona mais detalhes e implementa o VQE usando o Qiskit.
1. O que é VQE?
O variational quantum eigensolver é um algoritmo que usa computação clássica e quântica em conjunto para realizar uma tarefa. Há 4 componentes principais em um cálculo de VQE:
- Um operador: Geralmente um Hamiltoniano, que chamaremos de , que descreve uma propriedade do seu sistema que você deseja otimizar. Outra forma de dizer isso é que você está buscando o autovetor desse operador que corresponde ao menor autovalor. Costumamos chamar esse autovetor de "estado fundamental".
- Um "ansatz" (palavra alemã que significa "abordagem"): é um circuito quântico que prepara um estado quântico que aproxima o autovetor que você está buscando. Na verdade, o ansatz é uma família de circuitos quânticos, porque alguns dos gates do ansatz são parametrizados, ou seja, recebem um parâmetro que podemos variar. Essa família de circuitos quânticos pode preparar uma família de estados quânticos que aproximam o estado fundamental.
- Um estimador: um meio de estimar o valor esperado do operador sobre o estado quântico variacional atual. Às vezes o que realmente nos interessa é simplesmente esse valor esperado, que chamamos de função de custo. Às vezes, nos importa uma função mais complicada que ainda pode ser escrita a partir de um ou mais valores esperados.
- Um otimizador clássico: um algoritmo que varia parâmetros tentando minimizar a função de custo.
Vamos olhar cada um desses componentes com mais profundidade.
1.1 O operador (Hamiltoniano)
No centro de um problema de VQE está um operador que descreve um sistema de interesse. Vamos assumir aqui que o menor autovalor e o autovetor correspondente desse operador são úteis para algum propósito científico ou de negócio. Exemplos podem incluir um Hamiltoniano químico descrevendo uma molécula, de forma que o menor autovalor do operador corresponda à energia do estado fundamental da molécula, e o autoestado correspondente descreva a geometria ou a configuração eletrônica da molécula. Ou o operador pode descrever o custo de um determinado processo a ser otimizado, e os autoestados podem corresponder a rotas ou práticas. Em algumas áreas, como a física, um "Hamiltoniano" quase sempre se refere a um operador que descreve a energia de um sistema físico. Mas em computação quântica, é comum ver operadores quânticos que descrevem um problema de negócio ou logístico também chamados de "Hamiltoniano". Vamos adotar essa convenção aqui.

Mapear um problema físico ou de otimização para qubits é geralmente uma tarefa não trivial, mas esses detalhes não são o foco deste curso. Uma discussão geral sobre como mapear um problema para um operador quântico pode ser encontrada em Quantum computing in practice. Uma análise mais detalhada do mapeamento de problemas de química para operadores quânticos pode ser encontrada em Quantum Chemistry with VQE.
Para os propósitos deste curso, vamos assumir que a forma do Hamiltoniano é conhecida. Por exemplo, um Hamiltoniano para uma molécula simples de hidrogênio (sob certas suposições de espaço ativo e usando o mapper Jordan-Wigner) é:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Note que no Hamiltoniano acima, há termos como ZZII e YYYY que não comutam entre si. Ou seja, para avaliar ZZII, precisaríamos medir o operador de Pauli Z no qubit 3 (entre outras medições). Mas para avaliar YYYY, precisamos medir o operador de Pauli Y nesse mesmo qubit, o qubit 3. Existe uma relação de incerteza entre os operadores Y e Z no mesmo qubit; não podemos medir ambos os operadores ao mesmo tempo. Vamos revisitar esse ponto mais abaixo, e de fato ao longo de todo o curso.
O Hamiltoniano acima é um operador matricial . Diagonalizar o operador para encontrar seu menor autovalor de energia não é difícil.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
Solucionadores clássicos de autovalores por força bruta não conseguem escalar para descrever as energias ou geometrias de sistemas de átomos muito grandes, como medicamentos ou proteínas. O VQE é uma das primeiras tentativas de aproveitar a computação quântica nesse problema.
Vamos encontrar Hamiltonianos nesta lição muito maiores do que o acima. Mas seria um desperdício forçar os limites do que o VQE pode fazer antes de apresentarmos algumas das ferramentas mais avançadas que podem complementar ou substituir o VQE, mais adiante neste curso.
1.2 Ansatz
A palavra "ansatz" é alemã e significa "abordagem". O plural correto em alemão é "ansätze", embora muitas vezes se veja "ansatzes" ou "ansatze". No contexto do VQE, um ansatz é o circuito quântico usado para criar uma função de onda de múltiplos qubits que mais se aproxima do estado fundamental do sistema que você está estudando, e que, portanto, produz o menor valor esperado do seu operador. Esse circuito quântico vai conter parâmetros variacionais (frequentemente reunidos no vetor de variáveis ).

Um conjunto inicial de valores dos parâmetros variacionais é escolhido. Vamos chamar a operação unitária do ansatz no circuito de . Por padrão, todos os qubits em computadores quânticos IBM® são inicializados no estado . Quando o circuito é executado, o estado dos qubits será
Se tudo que precisássemos fosse a menor energia (usando a linguagem dos sistemas físicos), poderíamos estimá-la simplesmente medindo a energia muitas vezes e tomando o menor valor. Mas tipicamente também queremos a configuração que produz essa menor energia ou autovalor. Então o próximo passo é a estimação do valor esperado do Hamiltoniano, que é obtida por meio de medições quânticas. Há muita coisa envolvida nisso. Mas podemos entender esse processo qualitativamente observando que a probabilidade de medir uma energia (novamente usando a linguagem dos sistemas físicos) está relacionada ao valor esperado por:
A probabilidade também está relacionada à sobreposição entre o autoestado e o estado atual do sistema :
Portanto, fazendo muitas medições dos operadores de Pauli que compõem nosso Hamiltoniano, podemos estimar o valor esperado do Hamiltoniano no estado atual do sistema . O próximo passo é variar os parâmetros e tentar se aproximar mais do estado fundamental (de menor energia) do sistema. Por causa dos parâmetros variacionais no ansatz, às vezes ele é chamado de forma variacional.
Antes de avançarmos para esse processo variacional, observe que geralmente é útil iniciar seu estado a partir de um estado de "boa estimativa inicial". Você pode saber o suficiente sobre seu sistema para fazer uma estimativa inicial melhor do que . Por exemplo, é comum inicializar os qubits no estado Hartree-Fock em aplicações químicas. Essa estimativa inicial, que não contém nenhum parâmetro variacional, é chamada de estado de referência. Vamos chamar o circuito quântico usado para criar o estado de referência de . Sempre que for importante distinguir o estado de referência do restante do ansatz, usamos: De forma equivalente:
1.3 Estimador
Precisamos de uma forma de estimar o valor esperado do nosso Hamiltoniano em um determinado estado variacional . Se pudéssemos medir diretamente o operador inteiro , isso seria tão simples quanto fazer muitas (digamos ) medições e calcular a média dos valores medidos:
Aqui, o símbolo nos lembra que esse valor esperado só seria exato no limite quando . Mas com milhares de medições sendo feitas em um circuito, o erro de amostragem do valor esperado é bastante baixo. Há outras considerações, como o ruído, que se tornam um problema para cálculos de precisão muito alta.
No entanto, geralmente não é possível medir de uma vez. pode conter múltiplos operadores de Pauli X, Y e Z não comutantes. Portanto, o Hamiltoniano deve ser dividido em grupos de operadores que podem ser medidos simultaneamente, e cada grupo deve ser estimado separadamente, combinando os resultados para obter um valor esperado. Vamos revisitar isso com mais detalhes na próxima lição, quando discutiremos a escalabilidade das abordagens clássicas e quânticas. Essa complexidade na medição é uma razão pela qual precisamos de código altamente eficiente para realizar essas estimativas. Nesta lição e além, usaremos o primitivo Estimator do Qiskit Runtime para esse propósito.
1.4 Otimizadores clássicos
Um otimizador clássico é qualquer algoritmo clássico projetado para encontrar extremos de uma função alvo (tipicamente um mínimo). Eles pesquisam o espaço de parâmetros possíveis em busca de um conjunto que minimize alguma função de interesse. Podem ser amplamente categorizados em métodos baseados em gradiente, que utilizam informações de gradiente, e métodos sem gradiente, que operam como otimizadores de caixa preta. A escolha do otimizador clássico pode impactar significativamente o desempenho de um algoritmo, especialmente na presença de ruído no hardware quântico. Otimizadores populares nessa área incluem Adam, AMSGrad e SPSA, que mostraram resultados promissores em ambientes com ruído. Otimizadores mais tradicionais incluem COBYLA e SLSQP.
Um fluxo de trabalho comum (demonstrado na Seção 3.3) é usar um desses algoritmos como o método dentro de um minimizador como a função minimize do scipy. Ela recebe como argumentos:
- Alguma função a ser minimizada. Geralmente é o valor esperado da energia. Mas essas funções são geralmente chamadas de "funções de custo".
- Um conjunto de parâmetros a partir dos quais iniciar a busca. Frequentemente chamado de ou .
- Argumentos, incluindo argumentos da função de custo. Em computação quântica com Qiskit, esses argumentos incluirão o ansatz, o Hamiltoniano e o estimador, que é discutido mais na próxima subseção.
- Um 'método' de minimização. Refere-se ao algoritmo específico usado para pesquisar o espaço de parâmetros. É aqui que especificaríamos, por exemplo, COBYLA ou SLSQP.
- Opções. As opções disponíveis podem diferir por método. Mas um exemplo que praticamente todos os métodos incluem é o número máximo de iterações do otimizador antes de encerrar a busca: 'maxiter'.

Em cada passo iterativo, o valor esperado do Hamiltoniano é estimado fazendo muitas medições. Essa energia estimada é retornada pela função de custo, e o minimizador atualiza as informações que tem sobre o cenário de energia. O que exatamente o otimizador faz para escolher o próximo passo varia de método para método. Alguns usam gradientes e selecionam a direção de maior descida. Outros podem levar o ruído em conta e podem exigir que o custo diminua por uma margem grande antes de aceitar que a energia verdadeira diminui naquela direção.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 O princípio variacional
Nesse contexto, o princípio variacional é muito importante; ele estabelece que nenhuma função de onda variacional pode produzir um valor esperado de energia (ou custo) menor do que o produzido pela função de onda do estado fundamental. Matematicamente,
Isso é fácil de verificar se notarmos que o conjunto de todos os autoestados de formam uma base completa para o espaço de Hilbert. Em outras palavras, qualquer estado e, em particular, pode ser escrito como uma soma ponderada (normalizada) desses autoestados de :
onde são constantes a serem determinadas, e . Deixamos isso como exercício para o leitor. Mas note a implicação: o estado variacional que produz o menor valor esperado de energia é a melhor estimativa do verdadeiro estado fundamental.
Teste seus conhecimentos
Leia a pergunta abaixo, pense na sua resposta e clique no triângulo para revelar a solução.
Verifique matematicamente que para qualquer estado variacional .
Resposta:
Usando a expansão dada do estado variacional em termos dos autoestados de energia,
podemos escrever o valor esperado de energia variacional como
Para todos os coeficientes . Portanto, podemos escrever
2. Comparação com o fluxo de trabalho clássico
Suponha que estamos interessados em uma matriz com N linhas e N colunas. Suponha que sua matriz seja tão grande que a diagonalização exata não é uma opção. Suponha ainda que você sabe o suficiente sobre o seu problema para fazer algumas suposições sobre a estrutura geral do autoestado alvo, e você quer explorar estados semelhantes à sua estimativa inicial para ver se o custo/energia pode ser reduzido ainda mais. Essa é uma abordagem variacional, e é um método usado quando a diagonalização exata não é uma opção.
2.1 Fluxo de trabalho clássico
Usando um computador clássico, isso funcionaria da seguinte forma:
- Faça uma estimativa de estado, com alguns parâmetros que você vai variar: . Embora essa estimativa inicial possa ser aleatória, isso não é aconselhável. Queremos usar o conhecimento do problema em questão para personalizar nossa estimativa o máximo possível.
- Calcule o valor esperado do operador com o sistema naquele estado:
- Altere os parâmetros variacionais e repita: .
- Use as informações acumuladas sobre o cenário de estados possíveis no seu subespaço variacional para fazer estimativas cada vez melhores e se aproximar do estado alvo. O princípio variacional garante que nosso estado variacional não pode produzir um autovalor menor do que o do estado fundamental alvo. Portanto, quanto menor o valor esperado, melhor é nossa aproximação do estado fundamental:
Vamos examinar a dificuldade de cada etapa nessa abordagem. Definir ou atualizar parâmetros é computacionalmente fácil; a dificuldade aí está em selecionar parâmetros iniciais úteis e fisicamente motivados. Usar informações acumuladas de iterações anteriores para atualizar parâmetros de forma que você se aproxime do estado fundamental é não trivial. Mas existem algoritmos de otimização clássica que fazem isso de forma bastante eficiente. Essa otimização clássica só é cara porque pode exigir muitas iterações; no pior caso, o número de iterações pode escalar exponencialmente com N. A etapa individual computacionalmente mais cara é quase certamente calcular o valor esperado da sua matriz usando um dado estado :
A matriz deve agir sobre o vetor de elementos, o que corresponde a: operações de multiplicação no pior caso. Isso deve ser feito a cada iteração de parâmetros. Para matrizes extremamente grandes, isso tem custo computacional elevado.
2.2 Fluxo de trabalho quântico e grupos de Paulis comutantes
Agora imagine transferir essa parte do cálculo para um computador quântico. Em vez de calcular esse valor esperado, você o estima preparando o estado no computador quântico usando seu ansatz variacional e então fazendo medições.
Isso pode parecer mais fácil do que realmente é. geralmente não é fácil de medir. Por exemplo, pode ser composto de muitos operadores de Pauli X, Y e Z não comutantes. Mas pode ser escrito como uma combinação linear de termos, , cada um dos quais é facilmente mensurável (por exemplo, operadores de Pauli ou grupos de operadores de Pauli comutantes qubit a qubit). O valor esperado de sobre algum estado é a soma ponderada dos valores esperados dos termos constituintes . Essa expressão vale para qualquer estado , mas vamos usá-la especificamente com nossos estados variacionais .
onde é uma string de Pauli como IZZX…XIYX, ou várias dessas strings que comutam entre si. Portanto, uma descrição do valor esperado que mais se aproxima das realidades da medição em computadores quânticos é
E no contexto da nossa função de onda variacional:
Cada um dos termos pode ser medido vezes, produzindo amostras de medição com e retornando um valor esperado e um desvio padrão . Podemos somar esses termos e propagar os erros pela soma para obter um valor esperado global e um desvio padrão .
Isso não requer multiplicação em grande escala, nem nenhum processo que necessite escalar como . Em vez disso, requer múltiplas medições no computador quântico. Se não forem necessárias muitas delas, essa abordagem pode ser eficiente. E é essa a parte quântica do VQE.
Mas vamos falar sobre as razões pelas quais isso pode não ser eficiente. Uma razão para muitas medições é reduzir a incerteza estatística nas suas estimativas, em cálculos de precisão muito alta. Outra razão é o número de strings de Pauli necessárias para abranger toda a sua matriz. Como as matrizes de Pauli (mais a identidade: X, Y, Z e I) abrangem o espaço de todos os operadores de uma dada dimensão, temos a garantia de que podemos escrever nossa matriz de interesse como uma soma ponderada de operadores de Pauli, como fizemos antes.
onde é uma string de Pauli atuando em todos os qubits que descrevem seu sistema como IZZX…XIYX, ou várias dessas strings que comutam entre si. Lembre-se de que o Qiskit usa a notação little endian, na qual o operador de Pauli a partir da direita atua no qubit. Portanto, podemos medir nosso operador medindo uma série de operadores de Pauli.
Mas não podemos medir todos esses operadores de Pauli simultaneamente. Operadores de Pauli (excluindo I) não comutam entre si se estiverem associados ao mesmo qubit. Por exemplo, podemos medir IZIZ e ZZXZ simultaneamente, porque podemos medir I e Z simultaneamente no terceiro qubit, e podemos conhecer I e X simultaneamente no primeiro qubit. Mas não podemos medir ZZZZ e ZZZX simultaneamente, porque Z e X não comutam, e ambos atuam no qubit 0. Leitores experientes podem se lembrar de que dois grupos de operadores de Pauli podem comutar como um conjunto, mesmo que as medições de cada qubit individualmente não comutem. O Estimator assume medições de Pauli em produto tensorial (via rotações de base), correspondendo ao agrupamento de operadores que são qubit-wise commuting. Portanto, para estimar simultaneamente duas strings (A e B) de operadores de Pauli usando o Estimator, os operadores de Pauli de cada qubit em A e B devem comutar. Isso significa que também não podemos medir ZZZZ e ZZXX simultaneamente.

Portanto, decompomoos nossa matriz em uma soma de Paulis atuando em diferentes qubits. Alguns elementos dessa soma podem ser medidos todos de uma vez; chamamos isso de grupo de Paulis comutantes. Dependendo de quantos termos não comutantes existem, podemos precisar de muitos desses grupos. Chame o número de tais grupos de strings de Pauli comutantes de . Se for pequeno, isso pode funcionar bem. Se tiver milhões de grupos, isso não será útil.
Os processos necessários para a estimação do valor esperado são reunidos no primitivo do Qiskit Runtime chamado Estimator. Para saber mais sobre o Estimator, consulte a referência da API na Documentação IBM Quantum®. Pode-se usar o Estimator diretamente, mas ele retorna muito mais do que apenas o menor autovalor de energia. Por exemplo, ele também retorna informações sobre o erro padrão do conjunto. Portanto, no contexto de problemas de minimização, frequentemente vemos o Estimator dentro de uma função de custo. Para saber mais sobre as entradas e saídas do Estimator, consulte este guia na Documentação IBM Quantum.
Você registra o valor esperado (ou a função de custo) para o conjunto de parâmetros usado no seu estado, e então atualiza os parâmetros. Com o tempo, você pode usar os valores esperados ou os valores da função de custo que estimou para aproximar um gradiente da sua função de custo no subespaço de estados amostrados pelo seu ansatz. Existem otimizadores clássicos tanto baseados em gradiente quanto sem gradiente. Ambos sofrem de possíveis problemas de treinabilidade, como múltiplos mínimos locais e grandes regiões do espaço de parâmetros com gradiente próximo de zero, chamadas de platôs estéreis (barren plateaus).

2.3 Fatores que determinam o custo computacional
O VQE não vai resolver todos os seus problemas mais difíceis de química quântica. Não mesmo. Mas ser melhor em todos os cálculos não é o objetivo. Mudamos o que determina o custo computacional.

Passamos de um processo cuja complexidade depende apenas da dimensão da matriz para um que depende da precisão exigida e do número de operadores de Pauli não comutantes que compõem a matriz. Essa última parte não tem análogo na computação clássica.
Com base nessas dependências, para matrizes esparsas ou matrizes envolvendo poucas strings de Pauli não comutantes, esse processo pode ser útil. Esse é o caso de sistemas de spins interagentes, por exemplo. Para matrizes densas, pode ser menos útil. Sabemos, por exemplo, que sistemas químicos frequentemente têm Hamiltonianos que envolvem centenas, milhares, até milhões de strings de Pauli. Há trabalhos interessantes sendo feitos para reduzir esse número de termos. Mas sistemas químicos podem ser mais adequados para alguns dos outros algoritmos que discutiremos neste curso.
Teste seus conhecimentos
Leia as perguntas abaixo, pense nas suas respostas e clique nos triângulos para revelar as soluções.
Considere um Hamiltoniano em quatro qubits que contém os termos:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Você quer ordenar esses termos em grupos de forma que todos os termos de um grupo possam ser medidos simultaneamente. Qual é o menor número de grupos que você pode formar de modo que todos os termos sejam incluídos?
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXXResposta:
É possível fazer em 4 grupos. Note que essas soluções tipicamente não são únicas.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
O que você espera que tipicamente dificulte a química quântica com VQE: o número de termos no Hamiltoniano ou encontrar um bom ansatz?
Resposta:
Na prática, existem ansätze altamente otimizados para contextos químicos. O número de termos no Hamiltoniano, e portanto o número de medições necessárias, costuma causar mais problemas.
3. Hamiltoniano de exemplo
Vamos colocar esse algoritmo em prática usando uma pequena matriz hamiltoniana para que possamos ver o que acontece em cada etapa. Vamos usar o framework de padrões do Qiskit:
-Etapa 1: Mapear o problema para circuitos quânticos e operadores -Etapa 2: Otimizar para o hardware alvo -Etapa 3: Executar no hardware alvo -Etapa 4: Pós-processar os resultados
3.1 Etapa 1: Mapear o problema para circuitos quânticos e operadores
Vamos usar o hamiltoniano definido acima, oriundo do contexto da química. Começamos com alguns imports gerais.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Novamente, assumimos que o hamiltoniano de interesse é conhecido. Vamos usar um hamiltoniano extremamente pequeno aqui, pois outros métodos discutidos neste curso serão mais eficientes para resolver problemas maiores.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
Há muitas opções de ansatz pré-fabricadas no Qiskit. Vamos usar efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
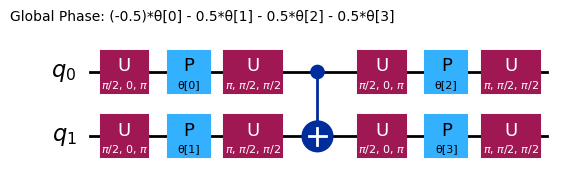
Diferentes ansätze terão estruturas de entrelaçamento e portas de rotação diferentes. O mostrado aqui usa portas CNOT para o entrelaçamento, e tanto portas Y quanto portas RZ parametrizadas para as rotações. Observe o tamanho desse espaço de parâmetros; isso significa que precisamos minimizar a função de custo em 4 variáveis (os parâmetros das portas RZ). Isso pode ser escalado, mas não indefinidamente. Executar um problema similar em 4 qubits, usando as 3 reps padrão do efficient_su2, resulta em 16 parâmetros variacionais.
3.2 Etapa 2: Otimizar para o hardware alvo
O ansatz foi escrito usando portas familiares, mas nosso circuito precisa ser transpilado para usar as portas base que podem ser implementadas em cada computador quântico. Selecionamos o backend menos ocupado.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Agora podemos transpilar nosso circuito para esse hardware e visualizar nosso ansatz transpilado.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Observe que as portas usadas mudaram, e os qubits do nosso circuito abstrato foram mapeados para qubits com numeração diferente no computador quântico. Precisamos mapear nosso hamiltoniano de forma idêntica para que nossos resultados sejam significativos.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Etapa 3: Executar no hardware alvo
3.3.1 Reportando os valores
Definimos aqui uma função de custo que recebe como argumentos as estruturas que construímos nas etapas anteriores: os parâmetros, o ansatz e o hamiltoniano. Ela também usa o estimador, que ainda não definimos. Incluímos código para rastrear o histórico da nossa função de custo, para que possamos verificar o comportamento de convergência.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
É muito vantajoso se você puder escolher os valores iniciais dos parâmetros com base no conhecimento do problema em questão e nas características do estado alvo. Não vamos assumir tal conhecimento e usaremos valores iniciais aleatórios.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Podemos ver os resultados brutos.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Etapa 4: Pós-processar os resultados
Se o procedimento terminar corretamente, então os valores no nosso dicionário devem ser iguais ao vetor solução e ao número total de avaliações da função, respectivamente. É fácil verificar isso:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

O IBM Quantum tem outros recursos de aprendizado relacionados ao VQE. Se você estiver pronto para colocar o VQE em prática, veja nosso tutorial: Estimativa de energia do estado fundamental da cadeia de Heisenberg com VQE. Se quiser mais informações sobre como criar hamiltonianos moleculares, veja esta lição no nosso curso sobre Química quântica com VQE. Se você tem interesse em entender mais a fundo como algoritmos variacionais como o VQE funcionam, recomendamos o curso Design de Algoritmos Variacionais.
Teste seu entendimento
Leia a pergunta abaixo, pense na sua resposta e clique no triângulo para revelar a solução.
Nesta seção, calculamos uma energia do estado fundamental a partir de um hamiltoniano. Se quiséssemos aplicar isso para determinar, por exemplo, a geometria de uma molécula, como faríamos essa extensão?
Resposta:
Precisaríamos introduzir variáveis para o espaçamento interatômico e os ângulos entre as ligações. Precisaríamos variar esses valores. Para cada variação, produziríamos um novo hamiltoniano (já que os operadores que descrevem a energia certamente dependem da geometria). Para cada hamiltoniano produzido e mapeado nos qubits, precisaríamos realizar uma otimização como a feita acima. De todos esses muitos problemas de otimização convergidos, a geometria que produziu a menor energia seria a adotada pela natureza. Isso é bem mais complexo do que o que foi mostrado acima. Esse cálculo é feito para a molécula mais simples, , aqui.
4. Relação do VQE com outros métodos
Nesta seção, vamos revisar as vantagens e desvantagens da abordagem original do VQE e apontar suas relações com outros algoritmos mais recentes.
4.1 Os pontos fortes e fracos do VQE
Alguns pontos fortes já foram mencionados. Entre eles:
- Adequação ao hardware moderno: Alguns algoritmos quânticos exigem taxas de erro muito menores, aproximando-se de tolerância a falhas em larga escala. O VQE não exige isso; ele pode ser implementado nos computadores quânticos atuais.
- Circuitos rasos: O VQE frequentemente emprega circuitos quânticos relativamente rasos. Isso torna o VQE menos suscetível a erros de porta acumulados e o torna adequado para muitas técnicas de mitigação de erros. Claro que os circuitos nem sempre são rasos; isso depende do ansatz utilizado.
- Versatilidade: O VQE pode (em princípio) ser aplicado a qualquer problema que possa ser formulado como um problema de autovalor/autovetor. Há muitas ressalvas que tornam o VQE impraticável ou desvantajoso para alguns problemas. Algumas delas são resumidas abaixo.
Algumas fraquezas do VQE e problemas para os quais ele é impraticável também foram descritos acima. Entre eles:
- Natureza heurística: O VQE não garante convergência para a energia correta do estado fundamental, pois seu desempenho depende da escolha do ansatz e dos métodos de otimização[1-2]. Se um ansatz inadequado for escolhido, sem o entrelaçamento necessário para o estado fundamental desejado, nenhum otimizador clássico conseguirá alcançar esse estado fundamental.
- Potencialmente muitos parâmetros: Um ansatz muito expressivo pode ter tantos parâmetros que as iterações de minimização se tornam muito demoradas.
- Alto custo de medição: No VQE, um estimador é usado para estimar o valor esperado de cada termo no hamiltoniano. A maioria dos hamiltonianos de interesse terá termos que não podem ser estimados simultaneamente. Isso pode tornar o VQE bastante custoso em recursos para sistemas grandes com hamiltonianos complicados[1].
- Efeitos do ruído: Quando o otimizador clássico está buscando um mínimo, cálculos ruidosos podem confundi-lo e desviá-lo do mínimo verdadeiro ou atrasar sua convergência. Uma possível solução para isso é aproveitar as técnicas de ponta de mitigação e supressão de erros[2-3] da IBM.
- Planaltos áridos (barren plateaus): Essas regiões de gradientes que desaparecem[2-3] existem mesmo na ausência de ruído, mas o ruído as torna mais problemáticas, pois a variação nos valores esperados devida ao ruído pode ser maior do que a variação causada pela atualização dos parâmetros nessas regiões.
4.2 Relação com outras abordagens
Adapt-VQE
O algoritmo ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver) é uma melhoria do algoritmo VQE original, projetado para aumentar a eficiência, a precisão e a escalabilidade para simulações quânticas, especialmente em química quântica.
O algoritmo VQE original descrito ao longo desta lição usa um ansatz predefinido e fixo para aproximar o estado fundamental do sistema. No nosso caso, usamos efficient_su2, com uma única repetição, usando portas de rotação Y e RZ. Embora os parâmetros nas portas RZ tenham mudado, a estrutura desse ansatz e as portas usadas não mudaram.
O ADAPT-VQE aborda as limitações do VQE por meio de construção adaptativa de ansatz. Em vez de começar com um ansatz fixo, o ADAPT-VQE constrói o ansatz dinamicamente de forma iterativa. Em cada etapa, ele seleciona o operador de um pool predefinido (como operadores de excitação fermiônica) que tem o maior gradiente em relação à energia. Isso garante que apenas os operadores mais impactantes sejam adicionados, resultando em um ansatz compacto e eficiente[4-6]. Essa abordagem pode ter vários efeitos benéficos:
- Profundidade de circuito reduzida: Ao desenvolver o ansatz de forma incremental e focando apenas nos operadores necessários, o ADAPT-VQE minimiza as operações de porta em comparação com as abordagens VQE tradicionais[5,7].
- Precisão aprimorada: A natureza adaptativa permite que o ADAPT-VQE recupere mais energia de correlação em cada etapa, tornando-o especialmente eficaz para sistemas fortemente correlacionados onde o VQE tradicional tem dificuldades[8,9].
- Escalabilidade e robustez ao ruído: O ansatz compacto reduz o acúmulo de erros de porta, diminui a sobrecarga computacional e limita o número de parâmetros variacionais que precisam ser minimizados.
O ADAPT-VQE ainda não é perfeito. Em alguns casos, pode ficar preso ou ser desacelerado por mínimos locais e pode sofrer de superparametrização. Ele também pode ser bastante custoso em recursos, pois exige o cálculo de gradientes e a otimização de parâmetros com muitas estruturas de portas.
Estimativa de fase quântica (QPE)
O QPE tem um propósito semelhante ao VQE, mas é muito diferente em implementação. O QPE exige computadores quânticos tolerantes a falhas devido aos seus circuitos quânticos geralmente profundos e ao alto nível de coerência que requer. Uma vez que o QPE possa ser implementado, ele seria mais preciso do que o VQE. Uma forma de descrever a diferença é por meio da precisão em função da profundidade do circuito. O QPE atinge precisão com profundidades de circuito escalando como [10]. O VQE requer amostras para atingir a mesma precisão[10,11].
Krylov, SQD, QSCI e outros neste curso
O VQE ajudou a estabelecer algoritmos quânticos que ainda dependem de computadores clássicos, não apenas para operar o computador quântico, mas para partes substanciais do algoritmo. Vários desses algoritmos são o foco do restante deste curso. Aqui, damos uma explicação superficial de alguns deles, apenas para compará-los e contrastá-los com o VQE. Eles serão explicados com muito mais detalhes nas lições subsequentes.
Diagonalização quântica de Krylov (KQD)
Os métodos de subespaço de Krylov são formas de projetar uma matriz em um subespaço para reduzir sua dimensão e torná-la mais gerenciável, mantendo as características mais importantes. Um truque nesse método é gerar um subespaço que preserve essas características; resulta que gerar esse subespaço está intimamente relacionado a um método bem estabelecido em computadores quânticos chamado Trotterização.
Há algumas variantes dos métodos de Krylov quântico, mas geralmente a abordagem é:
- Usar o computador quântico para gerar um subespaço (o subespaço de Krylov) por meio da Trotterização
- Projetar a matriz de interesse nesse subespaço de Krylov
- Diagonalizar o novo hamiltoniano projetado usando um computador clássico
Diagonalização quântica baseada em amostragem (SQD)
A diagonalização quântica baseada em amostragem (SQD) está relacionada ao método de Krylov no sentido de que também tenta reduzir a dimensão de uma matriz a ser diagonalizada enquanto preserva características essenciais. O SQD faz isso da seguinte forma:
- Começa com um chute inicial para o estado fundamental e prepara o sistema nesse estado fundamental.
- Usa o Sampler para amostrar as bitstrings que compõem esse estado.
- Usa a coleção de estados da base computacional do sampler como o subespaço no qual você projeta sua matriz de interesse.
- Diagonaliza a matriz menor e projetada usando um computador clássico.
Isso está relacionado ao VQE no sentido de que aproveita a computação clássica e quântica para componentes substanciais do algoritmo. Ambos também compartilham a exigência de que preparemos um bom chute inicial ou ansatz. Mas a distribuição do trabalho entre os computadores clássico e quântico no SQD se assemelha mais à do método de Krylov.
Na verdade, o método de Krylov e o SQD foram combinados recentemente no método de diagonalização quântica de Krylov baseada em amostragem (SKQD) [12].
Interação de configuração de subespaço quântico
A Interação de Configuração Selecionada Quânticamente (QSCI)[13] é um algoritmo que produz um estado fundamental aproximado de um hamiltoniano amostrando uma função de onda de teste para identificar os estados da base computacional mais significativos, a fim de gerar um subespaço para uma diagonalização clássica.
Tanto o SQD quanto o QSCI usam um computador quântico para construir um subespaço reduzido. O ponto forte adicional do QSCI está na sua preparação de estado, especialmente no contexto de problemas de química. Ele aproveita várias estratégias, como o uso de estados evoluídos no tempo [14] e um conjunto de ansätze inspirados em química. Ao focar na preparação eficiente de estado, o QSCI reduz os custos computacionais quânticos para hamiltonianos químicos, mantendo alta fidelidade e aproveitando a robustez ao ruído das técnicas de amostragem de estados quânticos [15]. O QSCI também oferece uma técnica de construção adaptativa que fornece mais ansätze para um resultado melhor.
O fluxo de trabalho padrão do QSCI para problemas de química é o seguinte:
- Construir o hamiltoniano molecular usando o software de sua preferência (como o SciPy).
- Preparar um algoritmo QSCI selecionando um estado inicial adequado e um ansatz inspirado em química com um conjunto de parâmetros pré-selecionados.
- Amostrar estados da base significativos e diagonalizar o hamiltoniano usando um computador clássico para obter a energia do estado fundamental.
- Frequentemente, usa-se recuperação de configuração [16] e pós-seleção de simetria [15] como técnica de pós-processamento.
- Opcionalmente, o fluxo de trabalho do QSCI adaptativo tem um loop de otimização adicional da etapa 2 à etapa 3, usando mais ansätze com estados iniciais aleatórios.
Teste seu entendimento
Leia as perguntas abaixo, pense nas suas respostas e clique nos triângulos para revelar as soluções.
O que o VQE tem em comum com todos os outros métodos listados acima (exceto o QPE, que não é descrito em grande detalhe)?
Resposta:
Todos envolvem um estado de teste ou função de onda de algum tipo. Todos funcionam melhor quando o chute inicial para esse estado de teste é excelente.
Outra resposta correta é que todos são mais fáceis de implementar quando o hamiltoniano é fácil de medir (pode ser agrupado em relativamente poucos grupos de operadores de Pauli comutantes).
O que o VQE tem em comum com nenhum dos outros métodos listados acima?
Resposta:
Otimizadores clássicos. Nenhum dos outros usa algoritmos de otimização clássicos para selecionar parâmetros variacionais.
Referências
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/