Diagonalização Quântica de Krylov
Nesta lição sobre diagonalização quântica de Krylov (DQK) vamos responder às seguintes perguntas:
- O que é o método de Krylov, de forma geral?
- Por que o método de Krylov funciona e em quais condições?
- Qual é o papel da computação quântica nesse processo?
A parte quântica dos cálculos é baseada principalmente no trabalho da Ref [1].
O vídeo abaixo fornece uma visão geral dos métodos de Krylov na computação clássica, motiva seu uso e explica como a computação quântica pode desempenhar um papel nesse fluxo de trabalho. O texto a seguir oferece mais detalhes e implementa um método de Krylov tanto de forma clássica quanto usando um computador quântico.
1. Introdução aos métodos de Krylov
Um método de subespaço de Krylov pode se referir a qualquer um dos vários métodos construídos em torno do que é chamado de subespaço de Krylov. Uma revisão completa deles está além do escopo desta lição, mas as Refs [2-4] podem oferecer substancialmente mais embasamento. Aqui, vamos nos concentrar no que é um subespaço de Krylov, como e por que ele é útil na resolução de problemas de autovalores, e, finalmente, como ele pode ser implementado em um computador quântico.
Definição: Dada uma matriz simétrica e semidefinida positiva , o espaço de Krylov de ordem é o espaço gerado pelos vetores obtidos multiplicando potências crescentes de uma matriz , até , por um vetor de referência .
Embora os vetores acima gerem o que chamamos de subespaço de Krylov, não há razão para acreditar que eles serão ortogonais entre si. Geralmente usa-se um processo iterativo de ortonormalização similar à ortogonalização de Gram-Schmidt. Aqui, o processo é ligeiramente diferente, pois cada novo vetor é tornado ortogonal aos demais à medida que é gerado. Nesse contexto, isso é chamado de iteração de Arnoldi. Começando com o vetor inicial , gera-se o próximo vetor , e então garante-se que esse segundo vetor seja ortogonal ao primeiro subtraindo sua projeção sobre . Ou seja:
Agora é fácil ver que pois
Fazemos o mesmo para o próximo vetor, garantindo que ele seja ortogonal aos dois anteriores:
Se repetirmos esse processo para todos os vetores, teremos uma base ortonormal completa para um espaço de Krylov. Note que o processo de ortogonalização aqui produzirá zero assim que , pois vetores ortogonais necessariamente geram o espaço completo. O processo também produzirá zero se algum vetor for um autovetor de , pois todos os vetores subsequentes serão múltiplos desse vetor.
1.1 Um exemplo simples: Krylov à mão
Vamos percorrer a geração de um subespaço de Krylov em uma matriz trivialmente pequena, para que possamos ver o processo. Começamos com uma matriz inicial de nosso interesse:
Para esse pequeno exemplo, podemos determinar os autovetores e autovalores facilmente até mesmo à mão. Mostramos aqui a solução numérica.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
Registramos aqui para comparação posterior:
Gostaríamos de estudar como esse processo funciona (ou falha) à medida que aumentamos a dimensão do nosso subespaço de Krylov, . Para isso, aplicaremos o seguinte processo:
- Gerar um subespaço do espaço vetorial completo começando com um vetor escolhido aleatoriamente (chame-o de se já estiver normalizado, como acima).
- Projetar a matriz completa sobre esse subespaço e encontrar os autovalores dessa matriz projetada .
- Aumentar o tamanho do subespaço gerando mais vetores, garantindo que sejam ortonormais, usando um processo similar à ortogonalização de Gram-Schmidt.
- Projetar sobre o subespaço maior e encontrar os autovalores da matriz resultante, .
- Repetir isso até que os autovalores convirjam (ou, nesse caso de brinquedo, até que você tenha gerado vetores que abrangem o espaço vetorial completo da matriz original ).
Uma implementação normal do método de Krylov não precisaria resolver o problema de autovalores para a matriz projetada em cada subespaço de Krylov à medida que ele é construído. Você poderia construir o subespaço da dimensão desejada, projetar a matriz sobre ele e diagonalizar a matriz projetada. Projetar e diagonalizar em cada dimensão de subespaço é feito apenas para verificar a convergência.
Dimensão :
Escolhemos um vetor aleatório, digamos
Se não estiver já normalizado, normalize-o.
Agora projetamos nossa matriz sobre o subespaço desse único vetor:
Essa é nossa projeção da matriz sobre o subespaço de Krylov quando ele contém apenas um único vetor, . O autovalor dessa matriz é trivialmente 4. Podemos pensar nisso como nossa estimativa de ordem zero dos autovalores (neste caso, apenas um) de . Embora seja uma estimativa ruim, tem a ordem de grandeza correta.
Dimensão :
Agora geramos o próximo vetor em nosso subespaço através da operação de sobre o vetor anterior:
Agora subtraímos a projeção desse vetor sobre nosso vetor anterior para garantir ortogonalidade.
Se não estiver já normalizado, normalize-o. Neste caso, o vetor já estava normalizado, então
Agora projetamos nossa matriz A sobre o subespaço desses dois vetores:
Ainda temos o problema de determinar os autovalores dessa matriz. Mas essa matriz é ligeiramente menor do que a matriz completa. Em problemas envolvendo matrizes muito grandes, trabalhar com esse subespaço menor pode ser altamente vantajoso.
Embora ainda não seja uma boa estimativa, é melhor do que a estimativa de ordem zero. Vamos executar mais uma iteração, para garantir que o processo esteja claro. No entanto, isso vai de encontro ao propósito do método, pois acabaremos diagonalizando uma matriz 3x3 na próxima iteração, o que significa que não economizamos tempo nem poder computacional.
Dimensão :
Agora geramos o próximo vetor em nosso subespaço através da operação de A sobre o vetor anterior:
Agora subtraímos a projeção desse vetor sobre os dois vetores anteriores para garantir ortogonalidade.
Se não estiver já normalizado, normalize-o. Neste caso, o vetor já estava normalizado, então
Agora projetamos nossa matriz sobre o subespaço desses vetores:
Agora determinamos os autovalores:
Esses autovalores são exatamente os autovalores da matriz original . Isso deve ser o caso, pois expandimos nosso subespaço de Krylov para abranger todo o espaço vetorial da matriz original .
Neste exemplo, o método de Krylov pode não parecer particularmente mais fácil do que a diagonalização direta. De fato, como veremos nas seções seguintes, o método de Krylov só é vantajoso acima de uma certa dimensão de matriz; este exemplo pretende nos ajudar a resolver problemas de autovalores/autovetores de matrizes extremamente grandes.
Este é o único exemplo que mostraremos trabalhado "à mão", mas a seção 2 abaixo mostra exemplos computacionais.
Esclarecimento de termos
Um equívoco comum é que existe apenas um único subespaço de Krylov para um dado problema. Mas, é claro, como há muitos vetores iniciais aos quais nossa matriz poderia ser aplicada, há muitos subespaços de Krylov possíveis. Usaremos apenas a expressão "o subespaço de Krylov" para nos referir a um subespaço de Krylov específico já definido para um exemplo específico. Para abordagens gerais de resolução de problemas, nos referiremos a "um subespaço de Krylov".
Um esclarecimento final é que é válido se referir a um "espaço de Krylov". Com frequência, vê-se chamado de "subespaço de Krylov" por causa de seu uso no contexto de projetar matrizes de um espaço inicial em um subespaço. Mantendo esse contexto, vamos nos referir principalmente a ele como subespaço aqui.
Teste seu entendimento
Leia as perguntas abaixo, pense na sua resposta e clique no triângulo para revelar cada solução.
Explique por que não é (a) útil e (b) possível estender a dimensão do subespaço de Krylov além da dimensão da matriz de interesse.
Resposta:
(a) Como estamos ortonormalizando os vetores à medida que os produzimos, um conjunto de tais vetores formará uma base completa, o que significa que uma combinação linear deles pode ser usada para criar qualquer vetor no espaço. (b) O processo de ortogonalização consiste em subtrair a projeção de um novo vetor sobre todos os vetores anteriores. Se todos os vetores anteriores abrangerem o espaço vetorial completo, subtrair as projeções sobre o subespaço completo sempre nos deixará com um vetor nulo.
Suponha que um colega pesquisador esteja demonstrando o método de Krylov aplicado a uma pequena matriz de brinquedo para alguns colegas. Esse pesquisador planeja usar a matriz e o vetor inicial:
e
Há algo errado com isso? Como você aconselharia seu colega?
Resposta:
Seu colega acidentalmente escolheu um autovetor como vetor inicial. Aplicar a matriz ao vetor inicial simplesmente retornará o mesmo vetor, escalado pelo autovalor. Isso não gerará um subespaço de dimensão crescente. Aconselhe seu colega a escolher um vetor inicial diferente, certificando-se de que não seja um autovetor.
Aplique o método de Krylov à matriz abaixo, selecionando um novo vetor inicial apropriado. Escreva as estimativas do autovalor mínimo na 0ª e na 1ª ordem do seu subespaço de Krylov.
Resposta:
Há muitas respostas possíveis dependendo da escolha do vetor inicial. Escolheremos:
Para obter , aplicamos uma vez a , e então tornamos ortogonal a
Na 0ª ordem, a projeção sobre o nosso subespaço de Krylov é
Na 1ª ordem, a projeção sobre esse subespaço de Krylov é
Isso pode ser feito à mão, mas é mais fácil usando numpy:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
saída:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
A estimativa do autovalor mínimo é -0,414.
1.2 Tipos de métodos de Krylov
"Métodos de subespaço de Krylov" pode se referir a qualquer uma das várias técnicas iterativas usadas para resolver grandes sistemas lineares e problemas de autovalores. O que todos têm em comum é que constroem uma solução aproximada a partir de um subespaço de Krylov
onde é o palpite inicial (veja Ref [5]). Eles diferem na forma como escolhem a melhor aproximação a partir desse subespaço, equilibrando fatores como taxa de convergência, uso de memória e custo computacional total. O foco desta lição é aproveitar a computação quântica no contexto dos métodos de subespaço de Krylov; uma discussão exaustiva desses métodos está além do seu escopo. As breves definições abaixo são apenas para contextualização e incluem algumas referências para aprofundamento nesses métodos.
O método do gradiente conjugado (GC): Esse método é usado para resolver sistemas lineares simétricos e definidos positivos[6]. Ele minimiza a norma-A do erro a cada iteração, tornando-o particularmente eficaz para sistemas que surgem de EDPs elípticas discretizadas[7]. Usaremos essa abordagem na próxima seção para motivar por que um subespaço de Krylov seria um subespaço eficaz para buscar soluções melhoradas de sistemas lineares.
O método dos resíduos mínimos generalizados (GMRES): Projetado para resolver sistemas lineares gerais não simétricos. Ele minimiza a norma do resíduo sobre um espaço de Krylov a cada iteração, tornando-o robusto, mas potencialmente com uso intensivo de memória para sistemas grandes[7].
O método dos resíduos mínimos (MINRES): Esse método é usado para resolver sistemas lineares simétricos indefinidos. É similar ao GMRES, mas aproveita a simetria da matriz para reduzir o custo computacional[8].
Outras abordagens notáveis incluem o método de ortogonalização completa (FOM), que está intimamente relacionado ao método de Arnoldi para problemas de autovalores, o método do gradiente bi-conjugado (BiCG) e o método de redução de dimensão induzida (IDR).
1.3 Por que o método do subespaço de Krylov funciona
Aqui vamos motivar que o método do subespaço de Krylov deve ser uma forma eficiente de aproximar os autovalores de uma matriz por meio de refinamento iterativo das aproximações de autovetores, através da perspectiva da descida mais íngreme. Vamos argumentar que, dado um palpite inicial para o estado fundamental, o espaço de correções sucessivas a esse palpite inicial que produz a convergência mais rápida é um subespaço de Krylov. Não chegamos a uma prova rigorosa do comportamento de convergência.
Assumimos que nossa matriz de interesse é simétrica e definida positiva. Isso torna nosso argumento mais relevante para o método GC acima. Não fazemos suposições sobre esparsidade aqui; tampouco afirmamos que deve ser Hermitiana (o que ela precisa ser se for um Hamiltoniano).
Tipicamente queremos resolver um problema da forma
Pode-se imaginar que onde é alguma constante, como em um problema de autovalores. Mas nosso enunciado do problema continua mais geral por ora.
Começamos com um vetor que é uma solução aproximada. Embora haja paralelos entre esse palpite e na Seção 1.1, não os aproveitamos aqui. Nosso palpite tem erro, que chamamos de
Também definimos o resíduo
Aqui usamos maiúsculo para distinguir o resíduo da dimensão do nosso subespaço de Krylov .

Agora queremos fazer um passo de correção da forma
que esperamos melhorar nossa aproximação. Aqui é algum vetor ainda a ser determinado. Seja o erro após a correção ser feita. Então

Estamos interessados em como nosso erro se comporta quando transformado pela nossa matriz. Então, vamos calcular a norma- do erro. Ou seja,
onde usamos a simetria de e também que Aqui é alguma constante independente de . Como mencionado na Seção 1.2, a norma- do erro não é a única quantidade que poderíamos escolher minimizar, mas é uma boa opção. Queremos ver como essa quantidade varia com nossa escolha de vetores de correção Então definimos a função estabelecendo
é simplesmente o erro como função da correção medido na norma-. Por isso, queremos escolher de forma que seja o menor possível. Para isso, calculamos o gradiente de . Usando a simetria de , temos
O gradiente aponta na direção de maior subida, o que significa que seu oposto nos dá a direção em que a função decresce mais: a direção da descida mais íngreme. No nosso palpite inicial , onde , temos que Assim, a função decresce mais na direção do resíduo Portanto, nossa escolha inicial se beneficiaria mais com a adição do vetor para algum escalar .
No próximo passo, escolhemos, novamente, um vetor e adicionamos seu valor à aproximação atual. Usando o mesmo argumento anterior, escolhemos para algum escalar . Continuamos dessa forma, de modo que a iteração do nosso vetor é
Equivalentemente, queremos construir o espaço do qual escolhemos nossas estimativas melhoradas adicionando , , e assim por diante, em ordem. O vetor estimado pertence a
Agora, usando a relação
vemos que
Ou seja, o espaço que construímos para aproximar de forma mais eficiente a solução correta é exatamente o espaço construído pela operação sucessiva da matriz sobre Um subespaço de Krylov é o espaço gerado pelos vetores de direções sucessivas de descida mais íngreme.
Por fim, reiteramos que não fizemos afirmações numéricas sobre o escalonamento dessa abordagem, nem discutimos o benefício comparativo para matrizes esparsas. Isso é apenas para motivar o uso de métodos de subespaço de Krylov e adicionar algum senso intuitivo a eles. Vamos agora explorar numericamente o comportamento desses métodos.
Teste seu entendimento
Leia a pergunta abaixo, pense na sua resposta e clique no triângulo para revelar a solução.
No fluxo de trabalho acima, propusemos minimizar a norma- do erro. Que outras quantidades alguém poderia considerar minimizar ao buscar o estado fundamental e seu autovalor?
Resposta:
Poderíamos imaginar usar o vetor resíduo em vez da norma- do erro. Pode haver casos em que considerar o vetor de erro em si seja útil.
2. Métodos de Krylov na computação clássica
Nesta seção, implementamos as iterações de Arnoldi computacionalmente para que possamos usar um subespaço de Krylov na resolução de problemas de autovalores. Vamos aplicar isso primeiro a um exemplo de pequena escala e, em seguida, examinar como o tempo de computação escala conforme o tamanho da matriz de interesse aumenta. Uma ideia-chave aqui é que a geração dos vetores que abrangem o espaço de Krylov será uma das maiores contribuições para o tempo total de computação necessário. A memória necessária varia entre os métodos de Krylov específicos. Mas restrições de memória podem limitar o uso de métodos de Krylov tradicionais.
2.1 Exemplo simples de pequena escala
No processo de criação de um subespaço de Krylov, precisamos ortonormalizar os vetores do nosso subespaço. Vamos definir uma função que recebe um vetor estabelecido do nosso subespaço vknown (não assumido como normalizado) e um vetor candidato para adicionar ao nosso subespaço vnext, tornando vnext ortogonal a vknown e normalizado. Vamos também definir uma função que percorre esse processo para todos os vetores estabelecidos no nosso subespaço de Krylov para garantir um conjunto totalmente ortonormal.
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
Agora vamos definir uma função que constrói um subespaço de Krylov cada vez maior de forma iterativa, até que o espaço dos vetores de Krylov abranja o espaço completo da matriz original. Isso nos permitirá ver quão bem os autovalores obtidos usando o método do subespaço de Krylov correspondem aos valores exatos, como função da dimensão do subespaço de Krylov. Importante: a nossa função krylov_full_build retorna os vetores de Krylov, os Hamiltonianos projetados, os autovalores e o tempo necessário.
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
Vamos testar isso em uma matriz que ainda é bastante pequena, mas maior do que gostaríamos de fazer manualmente.
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
Podemos verificar nossas funções garantindo que na última etapa (quando o espaço de Krylov é o espaço vetorial completo da matriz original) os autovalores do método de Krylov correspondam exatamente aos da diagonalização numérica exata:
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
Isso foi bem-sucedido. Claro, o que realmente importa é quão boa é nossa aproximação como função da dimensão do subespaço de Krylov. Como frequentemente nos preocupamos em encontrar estados fundamentais e outros autovalores mínimos (e por outras razões mais algébricas explicadas abaixo), vamos olhar nossa estimativa do menor autovalor como função da dimensão do subespaço de Krylov. Isso é
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
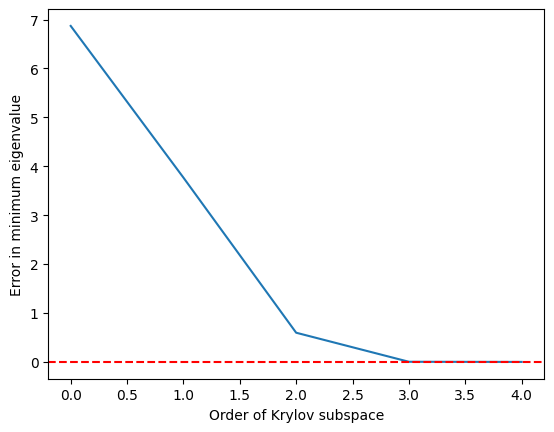
Vemos que o autovalor mínimo é atingido com bastante precisão quando o subespaço de Krylov cresce para e é perfeito a partir de
2.2 Escala de tempo com a dimensão da matriz
Vamos nos convencer de que o método de Krylov pode ser vantajoso em relação a eigensolvers numéricos exatos da seguinte forma:
- Construir matrizes aleatórias (não esparsas, não a aplicação ideal para KQD)
- Determinar autovalores usando dois métodos: diretamente com NumPy e usando um subespaço de Krylov.
- Escolhemos um limiar de precisão para nossos autovalores, antes de aceitarmos as estimativas de Krylov.
- Comparar o tempo de parede necessário para resolver das duas formas.
Ressalvas: Como discutiremos em detalhes abaixo, a diagonalização quântica de Krylov é mais bem aplicada a operadores cujas representações matriciais são esparsas e/ou podem ser escritas usando um número pequeno de grupos de operadores de Pauli comutativos. As matrizes aleatórias que estamos usando aqui não se encaixam nessa descrição. Elas são úteis apenas para investigar a escala em que os métodos clássicos de Krylov podem ser úteis. Em segundo lugar, ao usar o método de Krylov, vamos calcular autovalores usando muitos subespaços de Krylov de tamanhos diferentes. Vamos registrar o tempo necessário para o subespaço de Krylov de dimensão mínima que atinge a precisão exigida para o autovalor do estado fundamental. Novamente, isso é um pouco diferente de resolver um problema intratável para eigensolvers exatos, já que estamos usando a solução exata para avaliar a dimensão necessária.
Começamos gerando nosso conjunto de matrizes aleatórias.
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
Agora diagonalizamos cada matriz diretamente, usando numpy. Calculamos o tempo necessário para a diagonalização para comparação posterior.
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
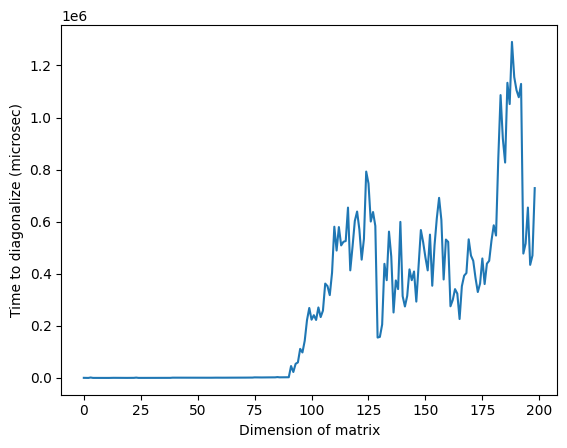
Note que na imagem acima, o tempo anormalmente alto em torno de uma dimensão de 125 pode ser devido à natureza aleatória das matrizes ou à implementação no processador clássico usado, mas não é reproduzível. Executar o código novamente produzirá um perfil diferente com picos anômalos diferentes.
Agora, para cada matriz, vamos construir um subespaço de Krylov e calcular autovalores em etapas. Em cada etapa, verificamos se o menor autovalor foi obtido dentro do nosso erro absoluto especificado. O subespaço que nos dá primeiro autovalores dentro do nosso erro especificado é o subespaço para o qual registraremos os tempos de computação. A execução desta célula pode levar vários minutos, dependendo da velocidade do processador. Fique à vontade para pular a avaliação ou reduzir a dimensão máxima das matrizes diagonalizadas. Olhar os resultados pré-calculados é suficiente.
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
Vamos plotar os tempos obtidos por esses dois métodos para comparação:
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

Esses são os tempos reais necessários, mas para fins de discussão, vamos suavizar essas curvas calculando a média sobre alguns pontos adjacentes / dimensões de matrizes. Isso é feito abaixo:
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

Note que o tempo necessário para a construção de um subespaço de Krylov inicialmente supera o tempo necessário para a diagonalização completa do numpy. Mas conforme o tamanho da matriz cresce, o método de Krylov se torna vantajoso. Isso vale mesmo se reduzirmos nosso erro aceitável, mas a vantagem aparece em um tamanho de matriz maior. Vale a pena analisar isso com cuidado.
A complexidade de tempo da diagonalização numérica é (com alguma variação entre algoritmos). A complexidade de tempo para gerar uma base ortonormal de vetores também é . Portanto, a vantagem do método de Krylov não está relacionada ao uso de base ortonormal, mas ao uso de uma base ortonormal particular que efetivamente seleciona os autovalores de interesse. Já vimos isso no esboço de uma prova na primeira seção desta lição, e isso é fundamental para as garantias de convergência nos métodos de Krylov. Vamos revisar nosso progresso até aqui:
- Para matrizes muito grandes, o método do subespaço de Krylov pode produzir autovalores aproximados dentro das tolerâncias exigidas mais rapidamente do que algoritmos de diagonalização tradicionais.
- Para essas matrizes muito grandes, a geração de um subespaço de Krylov é a parte mais demorada do método do subespaço de Krylov.
- Portanto, uma forma eficiente de gerar um subespaço de Krylov seria extremamente valiosa. É aqui finalmente que o computador quântico entra em cena.
Verifique sua compreensão
Leia a pergunta abaixo, pense na sua resposta e clique no triângulo para revelar a solução.
Consulte o gráfico suavizado de tempos de diagonalização vs. dimensão da matriz acima.
(a) Aproximadamente em qual dimensão de matriz o método de Krylov se tornou mais rápido, de acordo com esse gráfico?
(b) Quais aspectos do cálculo poderiam mudar a dimensão na qual o método de Krylov se torna mais rápido?
Resposta:
(a) As respostas podem variar se você executar o cálculo novamente, mas o método de Krylov se torna mais rápido aproximadamente na dimensão 80-85. (b) Há muitas respostas possíveis. Alguns fatores importantes são a precisão que exigimos e a esparsidade das matrizes sendo diagonalizadas.
3. Krylov via evolução temporal
Tudo o que descrevemos até agora pode ser feito classicamente. Então como e quando usaríamos um computador quântico? Para matrizes muito grandes, o método de Krylov pode exigir longos tempos de computação e grandes quantidades de memória. O tempo necessário para a operação matricial de em escala como no pior caso. Mesmo multiplicar matrizes esparsas por um vetor (o caso típico para resolvedores de Krylov clássicos) tem uma complexidade de tempo que escala como . Isso é feito para cada vetor que queremos no nosso subespaço. A dimensão do subespaço geralmente não é uma fração significativa de , e frequentemente escala como . Portanto, gerar todos os vetores escala como no pior caso. Embora haja outros passos, como a ortogonalização, esse é o escalonamento dominante a se ter em mente.
A computação quântica nos permite mudar quais atributos do problema determinam o escalonamento do tempo e dos recursos necessários. Em vez de dependência do tamanho da matriz em todo lugar, veremos coisas como número de shots e número de termos de Pauli não-comutativos que compõem o Hamiltoniano. Vamos explorar como isso funciona.
3.1 Evolução temporal
Lembre-se de que o operador que evolui temporalmente um estado quântico é (e é muito comum, especialmente em computação quântica, omitir o da notação). Uma forma de entender e até realizar essa função exponencial de um operador é olhar para a expansão em série de Taylor. Note que essa operação agindo sobre algum vetor inicial produz uma soma de termos com potências crescentes de aplicadas ao estado inicial. Parece que podemos simplesmente construir nosso subespaço de Krylov evoluindo temporalmente nosso estado de chute inicial!
O problema está em realizar a evolução temporal em um computador quântico real. Muitos dos termos no Hamiltoniano não comutam entre si. Portanto, embora alguns operadores exponenciais simples como correspondam a circuitos simples, Hamiltonianos gerais não. E como contêm termos não-comutativos, não podemos simplesmente decompor o exponencial em um produto de exponenciais simples, como podemos fazer com números.
Portanto, isso não é trivial, mas é um processo bem estudado em computação quântica. Realizamos a evolução temporal em computadores quânticos usando um processo chamado trotterização, que por si só é um assunto rico[10]. Mas em um nível bem alto, ao dividir a evolução temporal em passos muito pequenos, digamos passos de tamanho , limitamos os efeitos da não-comutatividade dos termos.
onde .
Vamos chamar de "subespaço de Krylov de potência" um subespaço de Krylov de ordem r que geramos no contexto clássico usando potências de H diretamente.
Agora geramos um espaço similar usando o operador unitário de evolução temporal ; vamos nos referir a isso como o "espaço de Krylov unitário" . O subespaço de Krylov de potência que usamos classicamente não pode ser gerado diretamente em um computador quântico, pois não é um operador unitário. Pode-se mostrar que usar o subespaço de Krylov unitário dá garantias de convergência similares às do subespaço de Krylov de potência, ou seja, o erro do estado fundamental converge eficientemente desde que o estado inicial tenha sobreposição com o verdadeiro estado fundamental que não seja exponencialmente pequena, e desde que haja uma lacuna suficiente entre autovalores. Veja a Ref [1] para uma discussão mais precisa sobre convergência.
Aqui, as potências de se tornam diferentes passos de tempo (a potência de é um avanço no tempo de ). Podemos rotular o elemento do subespaço que evolui temporalmente por um tempo total como .
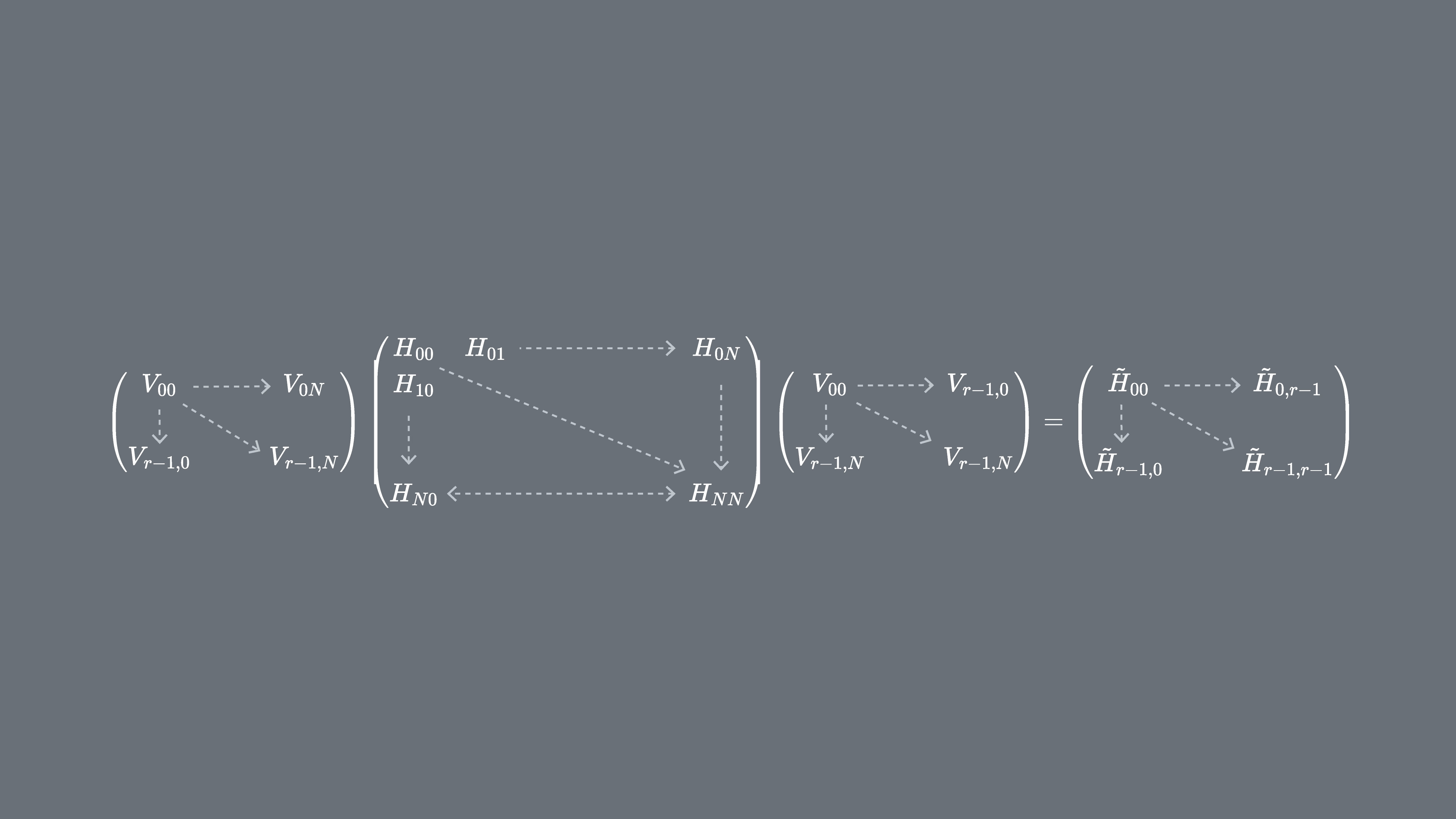
Podemos projetar nosso Hamiltoniano H no subespaço de Krylov unitário, . Em outras palavras, calculamos cada elemento de matriz de na base . Vamos nos referir a essa matriz projetada como .
3.2 Como implementar em um computador quântico
Os elementos de matriz de são dados pelos valores esperados , que podem ser estimados usando o computador quântico. Lembre-se de que pode ser escrito como uma soma de operadores de Pauli em diferentes qubits, e que nem todos os operadores de Pauli podem ser medidos simultaneamente. Podemos ordenar os termos de Pauli em grupos de termos comutativos e medir todos de uma vez. Mas podemos precisar de muitos desses grupos para cobrir todos os termos. Portanto, o número de grupos comutativos distintos nos quais os termos podem ser particionados, , torna-se importante.
Aqui é uma string de Pauli da forma ou um conjunto de tais strings de Pauli que comutam entre si. Dado que podemos escrever como tal soma de operadores mensuráveis, as seguintes expressões para os elementos de matriz de podem ser realizadas usando o primitivo Estimator do Qiskit Runtime.
Onde são os vetores do espaço de Krylov unitário e são os múltiplos do passo de tempo escolhido. Em um computador quântico, o cálculo de cada elemento de matriz pode ser feito com qualquer algoritmo que nos permita obter a sobreposição entre estados quânticos. Nesta lição vamos focar no teste de Hadamard. Dado que tem dimensão , o Hamiltoniano projetado no subespaço terá dimensões . Com suficientemente pequeno (geralmente é suficiente para obter convergência das estimativas dos autovalores), podemos então facilmente diagonalizar o Hamiltoniano projetado classicamente. No entanto, não podemos diagonalizar diretamente por causa da não-ortogonalidade dos vetores do espaço de Krylov. Teremos que medir suas sobreposições e construir uma matriz
Isso nos permite resolver o problema de autovalores em um espaço não-ortogonal (também chamado de problema de autovalores generalizado)
Pode-se então obter estimativas dos autovalores e autoestados de analisando as soluções desse problema de autovalores generalizado. Por exemplo, a estimativa da energia do estado fundamental é obtida tomando o menor autovalor e o estado fundamental do autovetor correspondente . Os coeficientes em determinam a contribuição dos diferentes vetores que abrangem .
Problema de autovalores generalizado
Por que não podemos simplesmente diagonalizar ? Como contém a informação sobre a geometria da base de Krylov (que é não-ortogonal em todos os casos exceto casos muito especiais), por si só não descreve uma projeção do Hamiltoniano completo, portanto seus autovalores não têm nenhuma relação particular com os do Hamiltoniano completo — eles poderiam ser quaisquer valores aleatórios. Resolver o problema de autovalores generalizado é necessário para obter os autovalores e autovetores aproximados correspondentes à projeção do Hamiltoniano completo no espaço de Krylov.
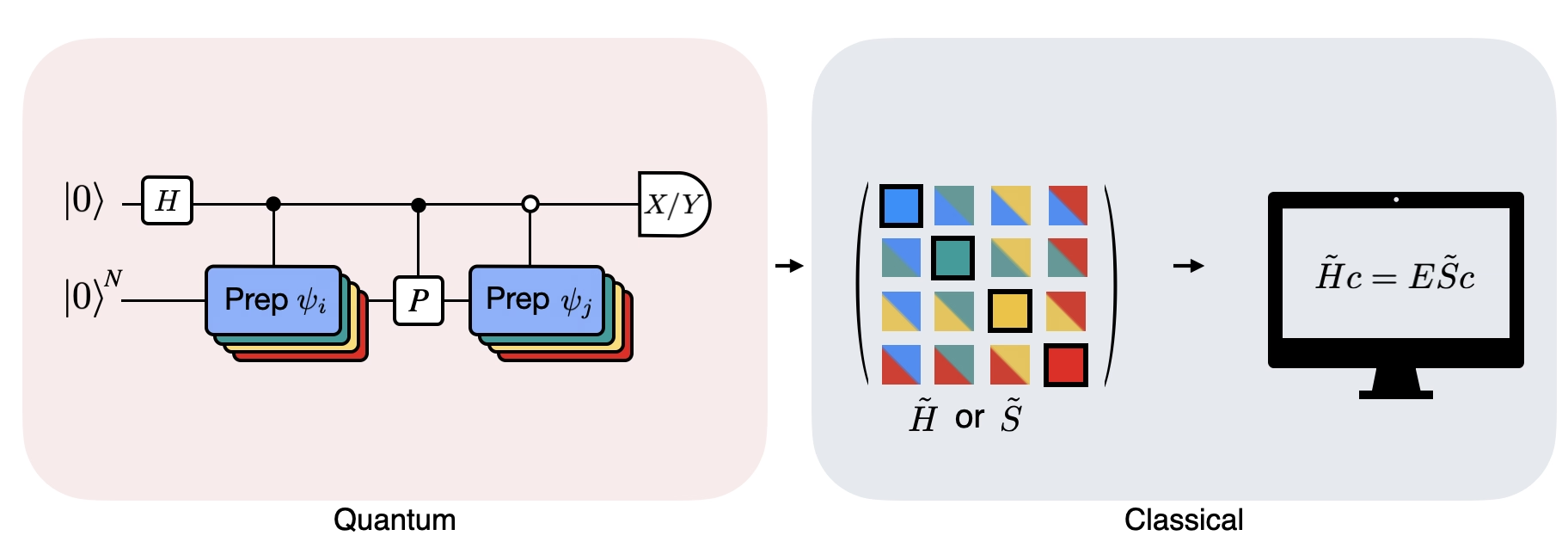
A figura mostra uma representação de circuito do teste de Hadamard modificado, um método usado para calcular a sobreposição entre diferentes estados quânticos. Para cada elemento de matriz , um teste de Hadamard entre os estados , é realizado. Isso é destacado na figura pelo esquema de cores para os elementos de matriz e as operações , correspondentes. Assim, um conjunto de testes de Hadamard para todas as combinações possíveis de vetores do espaço de Krylov é necessário para calcular todos os elementos de matriz do Hamiltoniano projetado . O fio superior no circuito do teste de Hadamard é um qubit ancila que é medido na base X ou Y; seu valor esperado determina o valor da sobreposição entre os estados. O fio inferior representa todos os qubits do Hamiltoniano do sistema. A operação prepara o qubit do sistema no estado controlado pelo estado do qubit ancila (similarmente para ) e a operação representa a decomposição de Pauli do Hamiltoniano do sistema . A implementação disso em um computador quântico é mostrada em mais detalhes abaixo.
4. Diagonalização quântica de Krylov num computador quântico
Agora vamos implementar a diagonalização quântica de Krylov num computador quântico de verdade. Vamos começar importando alguns pacotes úteis.
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
Definimos a função abaixo para resolver o problema de autovalor generalizado que acabamos de explicar.
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
Pelo menos nos benchmarks iniciais, é útil ter uma solução clássica exata para verificar o comportamento de convergência. A função abaixo calcula a energia do estado fundamental de um Hamiltoniano, usando o Hamiltoniano e o número de qubits como argumentos.
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 Passo 1: Mapear o problema para circuitos e operadores quânticos
Agora vamos definir um Hamiltoniano. Isso é diferente da função acima: aquela recebe um Hamiltoniano como argumento e retorna apenas o estado fundamental de forma clássica. O Hamiltoniano que definimos aqui determina os níveis de energia de todos os autoestados de energia, e ele pode ser construído usando operadores de Pauli e implementado num computador quântico.
Escolhemos um Hamiltoniano correspondente a uma cadeia de spins que podem ter qualquer orientação no espaço, chamada de "cadeia de Heisenberg". Assumimos que o -ésimo spin pode ser influenciado pelos seus vizinhos mais próximos (os spins -ésimo e -ésimo), mas não por vizinhos mais distantes. Também permitimos a possibilidade de que a interação entre os spins seja diferente quando eles apontam ao longo de eixos diferentes. Essa assimetria surge às vezes, por exemplo, devido à estrutura da rede cristalina na qual os spins estão inseridos.
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
O código abaixo restringe o Hamiltoniano aos estados de partícula única e usa a norma espectral para definir um bom tamanho para o nosso passo de tempo . Escolhemos heuristicamente um valor para o passo de tempo dt (baseado em limites superiores da norma do Hamiltoniano). A ref. [9] mostrou que um passo de tempo suficientemente pequeno é , e que é preferível, até certo ponto, subestimar esse valor em vez de superestimá-lo, pois superestimá-lo pode permitir que contribuições de estados de alta energia contaminem até mesmo o estado ótimo no espaço de Krylov. Por outro lado, escolher muito pequeno piora o condicionamento do subespaço de Krylov, já que os vetores de base de Krylov diferem menos de um passo de tempo para o outro.
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
Vamos especificar a dimensão máxima de Krylov que queremos usar, embora verifiquemos a convergência em dimensões menores. Também especificamos o número de passos de Trotter a usar na evolução temporal. Para fins desta lição, vamos escolher uma dimensão de Krylov pequena de apenas 4. Isso é bem limitado no contexto de aplicações realistas, mas é suficiente para este exemplo. Vamos explorar métodos em lições futuras que nos permitem escalar e projetar nossos Hamiltonianos em subespaços maiores.
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
Preparação de estado
Escolha um estado de referência que tenha alguma sobreposição com o estado fundamental. Para este Hamiltoniano, usamos um estado com uma excitação no qubit central como nosso estado de referência.
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

Evolução temporal
Podemos realizar o operador de evolução temporal gerado por um dado Hamiltoniano: via a aproximação de Lie-Trotter. Por simplicidade, usamos o PauliEvolutionGate integrado no circuito de evolução temporal. A sintaxe geral para isso é a seguinte.
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
Vamos usar uma versão disso abaixo no teste de Hadamard, avançando por tempos .
Teste de Hadamard
Lembre que queremos calcular os elementos de matriz tanto de quanto da matriz de Gram usando o teste de Hadamard. Vamos revisar como isso funciona neste contexto, focando primeiro na construção de O processo geral é representado graficamente abaixo. As camadas de blocos coloridos de preparação de estado servem de lembrete de que esse processo é realizado para todas as combinações de e no nosso subespaço.
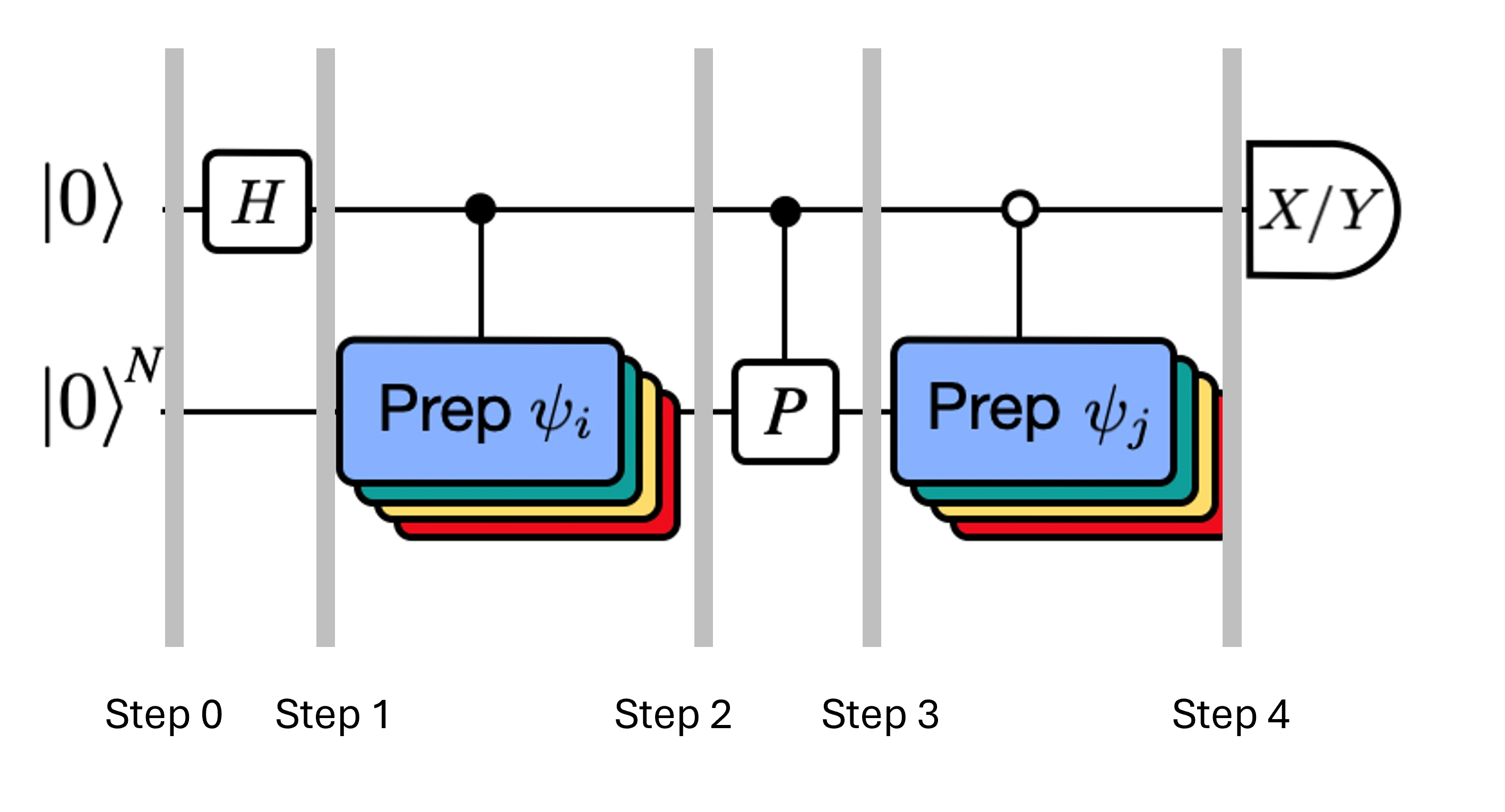
Os estados do sistema nas etapas indicadas são:
Aqui é um termo de Pauli na decomposição do Hamiltoniano (note que não pode ser uma combinação linear de múltiplos termos de Pauli comutantes, pois isso não seria unitário — o agrupamento é possível usando uma construção diferente que mostraremos mais adiante). , são operações controladas que preparam os vetores , do espaço de Krylov unitário, com . Aplicar medições de e a este circuito calcula as partes real e imaginária, respectivamente, dos elementos de matriz que precisamos.
A partir do Passo 4 acima, aplicamos a porta Hadamard ao qubit zero.
Em seguida, medimos ou .
Da identidade . De forma similar, medir resulta em
Adicionando esses passos à evolução temporal que configuramos anteriormente, escrevemos o seguinte.
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

Já alertamos sobre a profundidade envolvida nos circuitos de Trotter. Realizar o teste de Hadamard nessas condições pode gerar um circuito ainda mais profundo, especialmente após a decomposição em portas nativas. Isso aumenta ainda mais quando levamos em conta a topologia do dispositivo. Portanto, antes de utilizar qualquer tempo no computador quântico, é uma boa ideia verificar a profundidade de operações de 2 qubits do nosso circuito.
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
Um circuito com essa profundidade não consegue retornar resultados utilizáveis em computadores quânticos modernos. Para construirmos e precisamos de uma abordagem melhor. É por isso que apresentamos o teste de Hadamard eficiente a seguir.
4.2 Passo 2. Otimizar circuitos e operadores para o hardware alvo
Teste de Hadamard eficiente
Podemos otimizar os circuitos profundos do teste de Hadamard que obtivemos introduzindo algumas aproximações e nos apoiando em certas suposições sobre o Hamiltoniano do modelo. Por exemplo, considere o seguinte circuito para o teste de Hadamard:
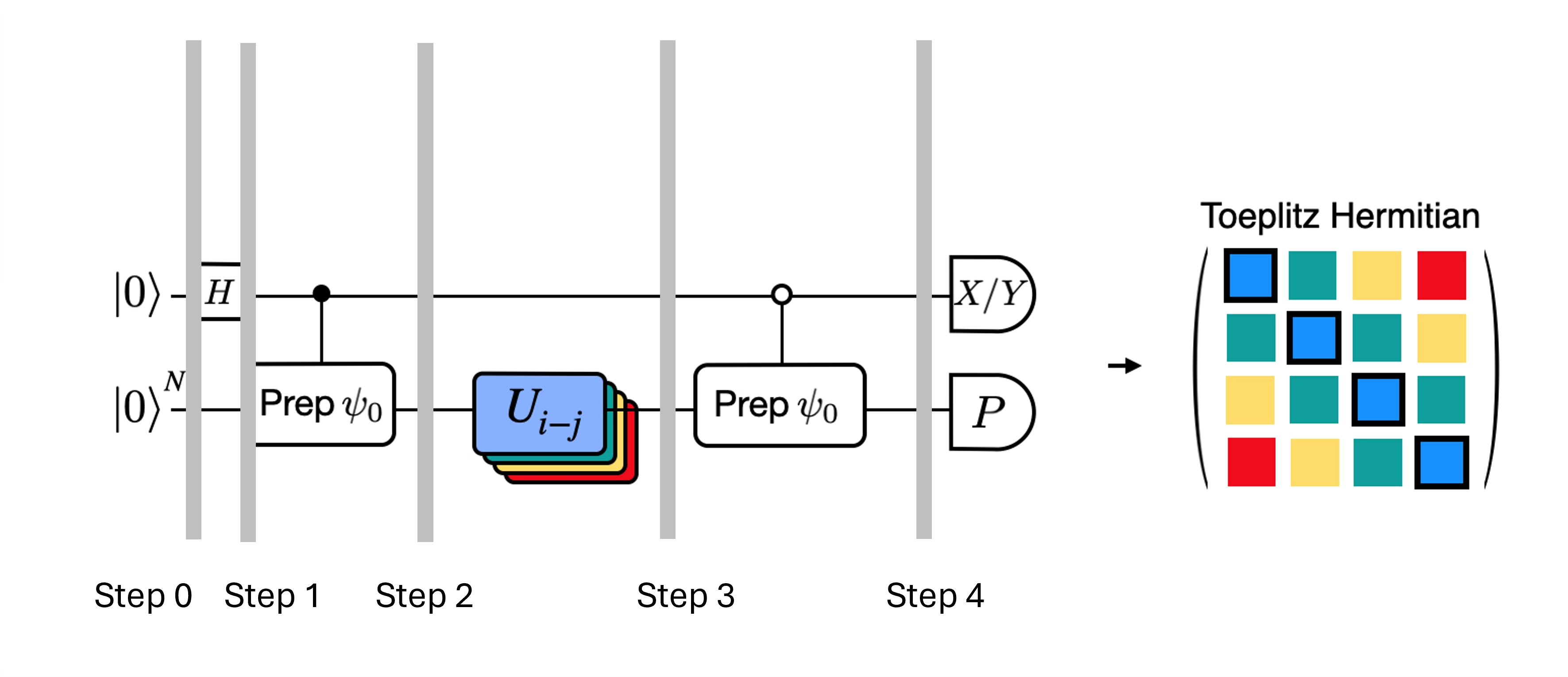
Suponha que podemos calcular classicamente , o autovalor de sob o Hamiltoniano . Isso é satisfeito quando o Hamiltoniano preserva a simetria U(1). Embora isso possa parecer uma suposição forte, há muitos casos em que é razoável assumir que existe um estado de vácuo (que nesse caso se mapeia ao estado ) que não é afetado pela ação do Hamiltoniano. Isso é verdade, por exemplo, para Hamiltonianos de química que descrevem moléculas estáveis (onde o número de elétrons é conservado). Dado que a porta prepara o estado de referência desejado , por exemplo, para preparar o estado HF em química, seria um produto de operações NOT de qubit único, portanto a controlada é simplesmente um produto de CNOTs. O circuito acima então implementa o seguinte estado antes da medição:
onde usamos o deslocamento de fase simulável classicamente do passo 2 para o passo 3. Portanto, os valores esperados são
Usando essas suposições, conseguimos escrever os valores esperados dos operadores de interesse com menos operações controladas. Na prática, só precisamos implementar a preparação de estado controlada e não evoluções temporais controladas. Reformular nosso cálculo dessa forma nos permite reduzir bastante a profundidade dos circuitos resultantes.
Observe que, como bônus, como o operador de Pauli agora aparece como uma medição ao final do circuito em vez de uma porta controlada no meio, ele pode ser medido junto com outros operadores de Pauli que comutam entre si, como na decomposição apresentada acima.
Decomposição do operador de evolução temporal com a decomposição de Trotter
Em vez de implementar o operador de evolução temporal de forma exata, podemos usar a decomposição de Trotter para implementar uma aproximação dele. Repetir várias vezes uma decomposição de Trotter de certa ordem nos dá uma redução adicional do erro introduzido pela aproximação. A seguir, construímos diretamente a implementação de Trotter da forma mais eficiente para o grafo de interação do Hamiltoniano que estamos considerando (interações apenas entre primeiros vizinhos). Na prática, inserimos rotações de Pauli , , com intensidades de acoplamento , e e um ângulo parametrizado , que correspondem à implementação aproximada de . Dada a diferença na definição das rotações de Pauli e da evolução temporal que queremos implementar, teremos que usar o parâmetro para obter uma evolução temporal de . Além disso, invertemos a ordem das operações para números ímpares de repetições dos passos de Trotter, o que é funcionalmente equivalente mas permite sintetizar operações adjacentes em um único unitário . Isso resulta em um circuito muito mais raso do que o obtido com a funcionalidade genérica PauliEvolutionGate().
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
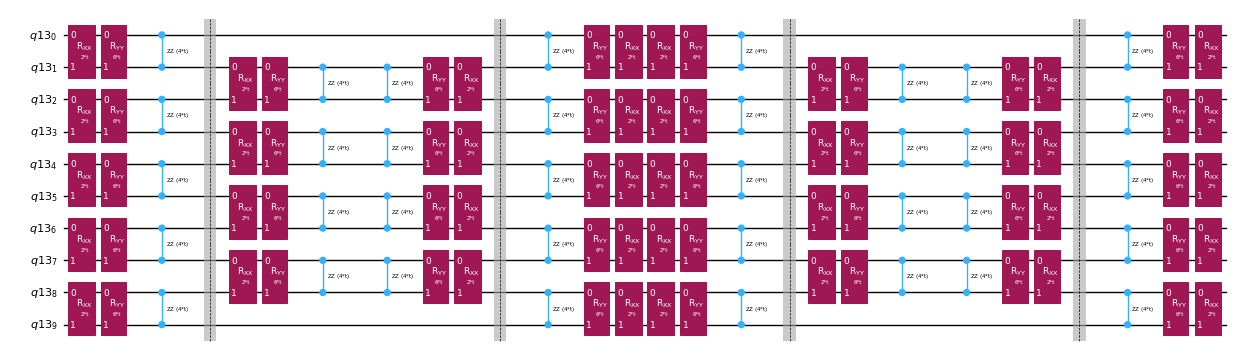
Preparamos novamente um estado inicial para esse teste de Hadamard eficiente.
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

Circuitos template para calcular os elementos de matriz de e via teste de Hadamard
A única diferença entre os circuitos usados no teste de Hadamard será a fase no operador de evolução temporal e os observáveis medidos. Portanto, podemos preparar um circuito template que representa o circuito genérico para o teste de Hadamard, com espaços reservados para as portas que dependem do operador de evolução temporal.
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
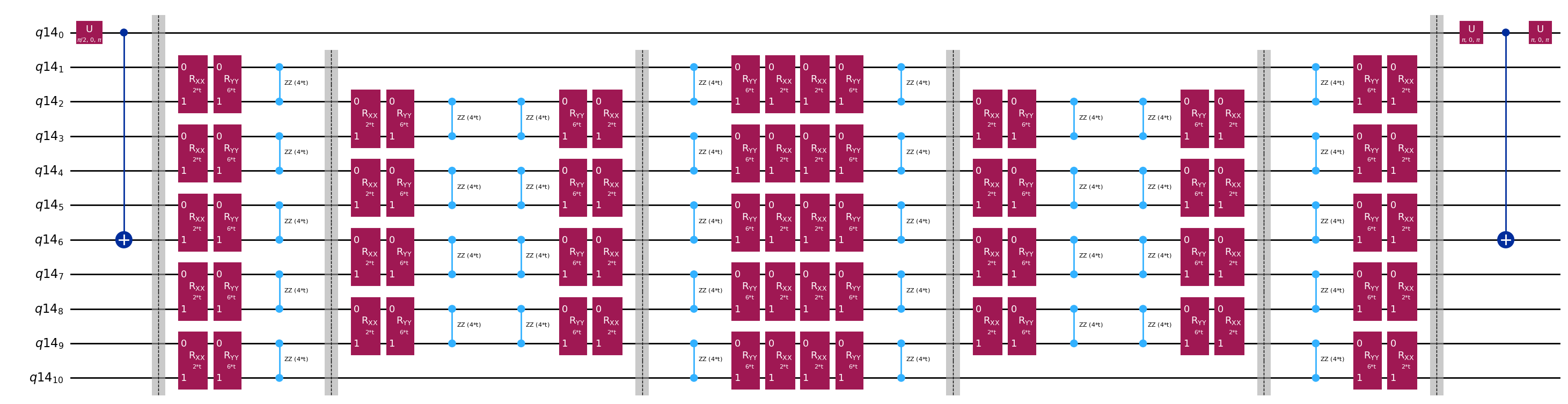
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
Essa profundidade é substancialmente menor em comparação com o teste de Hadamard original. Essa profundidade é gerenciável em computadores quânticos modernos, embora ainda seja bastante alta. Precisaremos usar mitigação de erros de última geração para obter resultados úteis.
Selecione um backend no qual executar nosso cálculo de Krylov quântico, para que possamos transpilar nosso circuito para rodar naquele computador quântico.
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
Agora transpilamos nossos circuitos e operadores.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

Após a otimização, a profundidade de dois qubits do circuito transpilado é reduzida ainda mais.
4.3 Passo 3. Executar usando uma primitiva do Qiskit Runtime
Agora criamos PUBs para execução com o Estimator.
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
Os circuitos para são calculáveis classicamente. Realizamos isso antes de passar para o caso usando um computador quântico.
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
Embora tenhamos conseguido reduzir a profundidade de portas em ordens de grandeza usando o teste de Hadamard eficiente, a profundidade ainda é suficiente para exigir mitigação de erros de última geração. Abaixo, especificamos os atributos da mitigação sendo usada. Todos os métodos utilizados são importantes, mas vale destacar especificamente a amplificação probabilística de erros (PEA). Essa técnica poderosa vem com um grande overhead quântico. O cálculo feito aqui pode levar 20 minutos ou mais para rodar em um computador quântico real. Você pode querer ajustar os parâmetros abaixo para aumentar ou reduzir a precisão e, consequentemente, o overhead. As configurações padrão abaixo produzem resultados de alta fidelidade.
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
Por fim, executamos os circuitos de e com o Estimator.
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 Passo 4. Pós-processar e analisar os resultados
O que obtivemos do computador quântico são os elementos individuais de matriz de e os grupos de Pauli que comutam entre si e compõem os elementos de matriz de . Esses termos precisam ser combinados para recuperar nossas matrizes, de modo que possamos resolver o problema de autovalor generalizado.
# Store the outputs as 'results'.
results = job.result()[0]
Calcular as matrizes do Hamiltoniano efetivo e de sobreposição
Primeiro, calcula a fase acumulada pelo estado durante a evolução temporal não controlada
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
Após obter os resultados das execuções dos circuitos, podemos pós-processar os dados para calcular os elementos de matriz de
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
E os elementos de matriz de
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
Por fim, podemos resolver o problema de autovalor generalizado para :
e obter uma estimativa da energia do estado fundamental
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
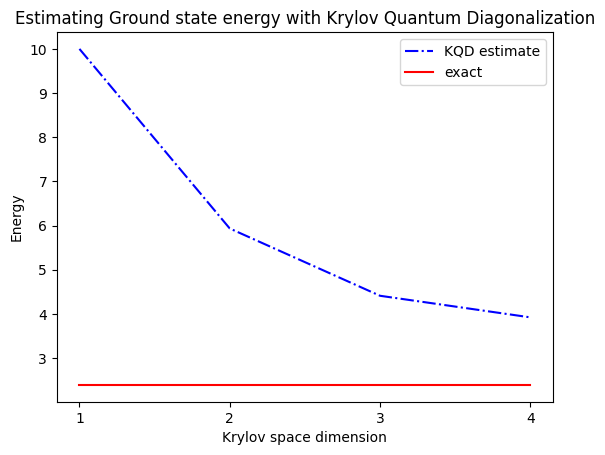
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
Para um setor de partícula única, podemos calcular eficientemente o estado fundamental desse setor do Hamiltoniano de forma clássica
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
5. Discussão e extensão
Para recapitular, começamos com um estado de referência, depois o evoluímos por diferentes períodos de tempo para gerar o subespaço de Krylov unitário. Projetamos nosso Hamiltoniano nesse subespaço. Também estimamos as sobreposições dos vetores do subespaço. Por fim, resolvemos classicamente o problema de autovalor generalizado de menor dimensão.
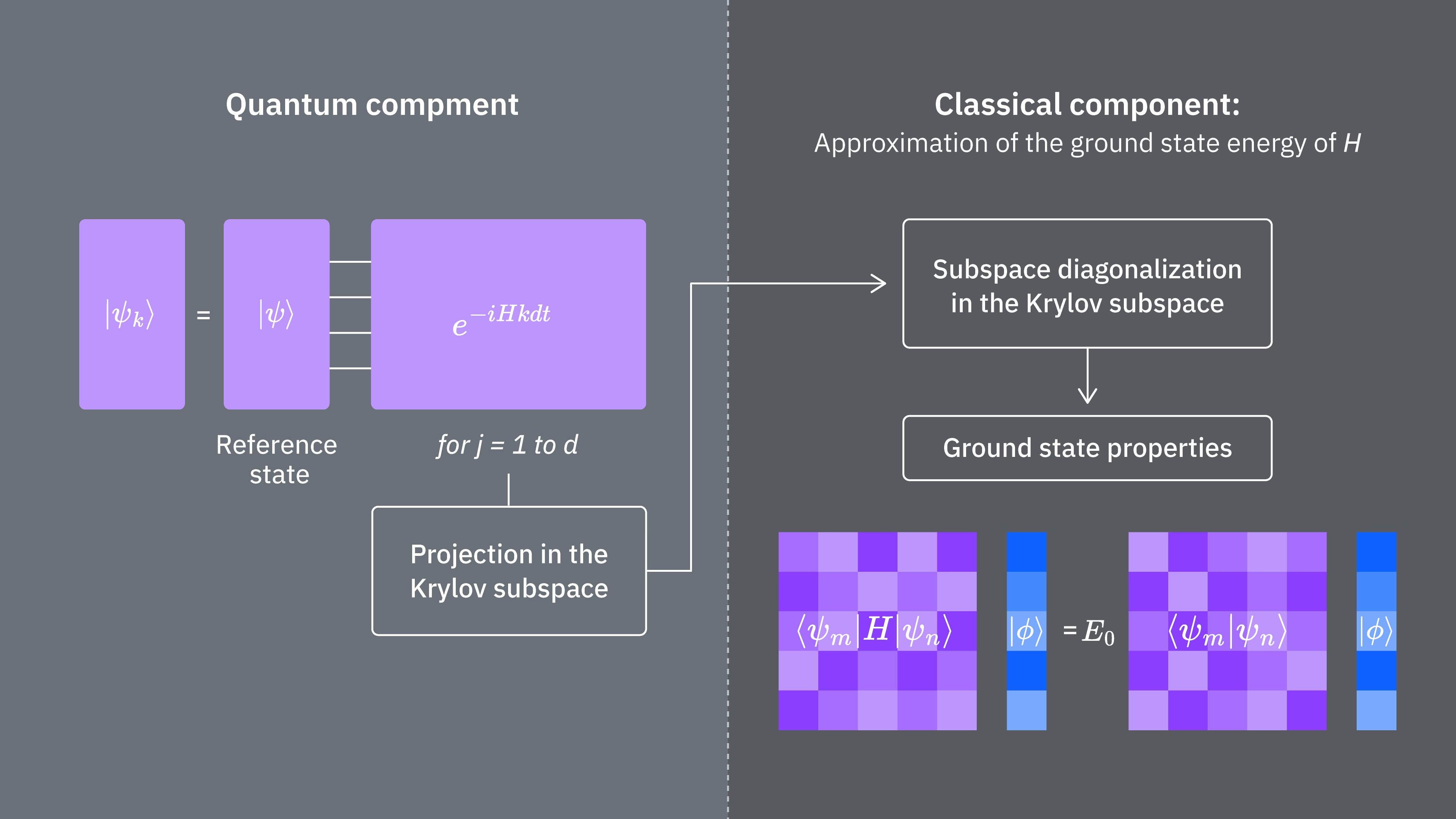
Vamos comparar o que determina os custos computacionais de usar a técnica de Krylov de forma clássica e quântica. Não há analogias perfeitas entre as abordagens clássica e quântica para todas as etapas. Esta tabela reúne alguns escalamentos de diferentes etapas para consideração.
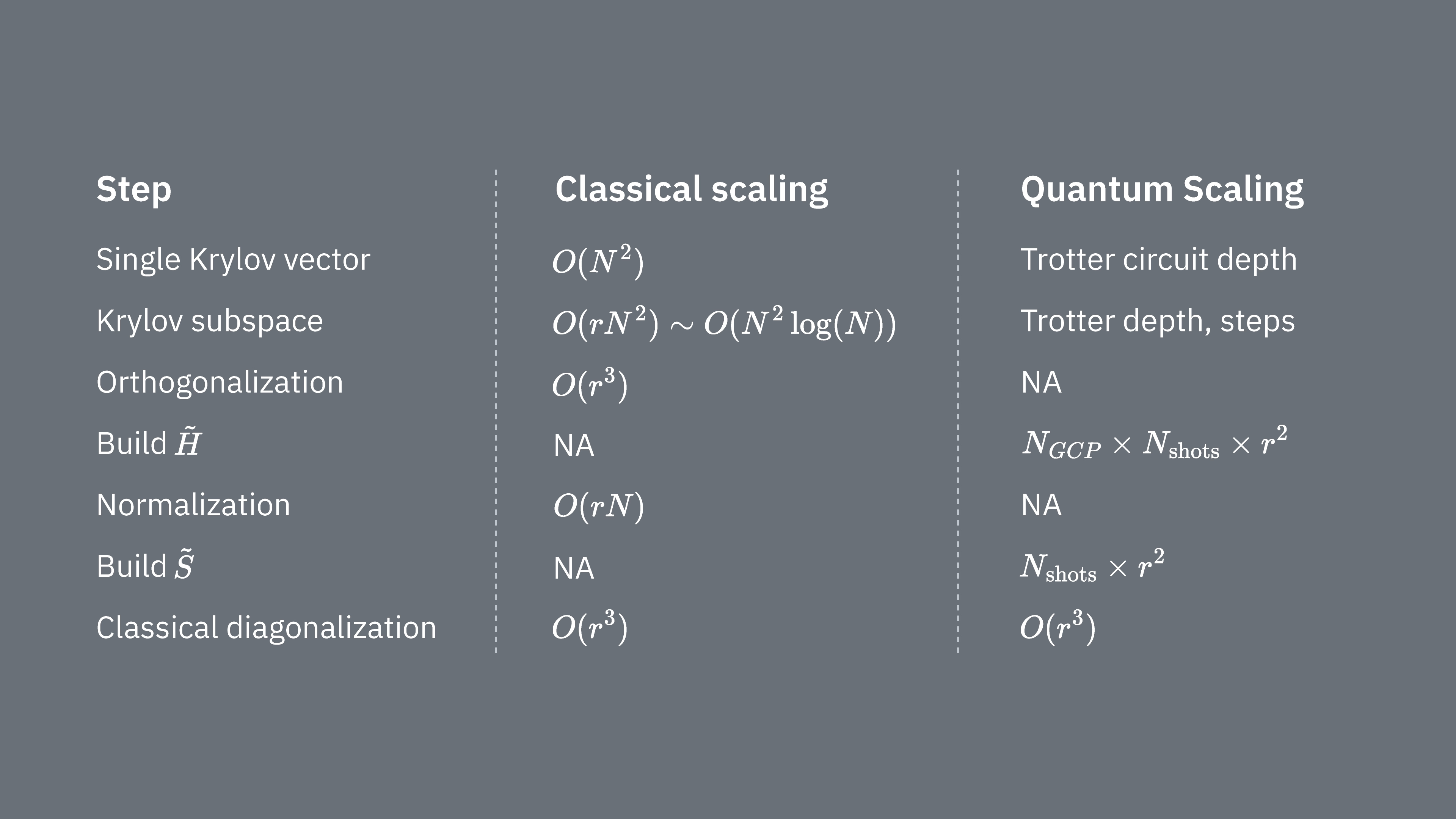
Lembre que os Hamiltonianos geralmente têm termos que não podem ser medidos simultaneamente (porque não comutam entre si). Agrupamos os termos do Hamiltoniano em grupos de operadores de Pauli que comutam e podem ser medidos ao mesmo tempo, e podem ser necessários muitos desses grupos para cobrir todos os termos que não comutam entre si. Construir em um computador quântico requer medições separadas para cada grupo de strings de Pauli que comutam no Hamiltoniano, e cada uma delas requer muitos shots. Precisamos fazer isso para elementos de matriz diferentes, correspondendo a combinações de diferentes fatores de evolução temporal. Às vezes há formas de reduzir isso, mas nesse tratamento aproximado, o tempo necessário escala como Os elementos de devem ser estimados, o que escala como . Por fim, resolver o problema de autovalor generalizado no espaço projetado, classicamente, tem custo
Vemos que a diagonalização de Krylov quântica pode ser útil nos casos em que o número de grupos de Pauli que comutam no Hamiltoniano é relativamente pequeno. Essas dependências de escalamento sugerem algumas aplicações onde o método de Krylov pode ser útil, e outras onde provavelmente não será. Alguns Hamiltonianos têm alta complexidade quando mapeados para qubits, envolvendo muitas strings de Pauli que não comutam e não podem ser facilmente particionadas em poucos grupos que comutam. Isso é frequentemente verdade para problemas de química quântica, por exemplo. Essa complexidade apresenta dois desafios principais para computadores quânticos de curto prazo:
- A estimativa de cada elemento de torna-se computacionalmente cara devido ao grande número de termos.
- Os circuitos de Trotter necessários tornam-se proibitivamente profundos.
Ambos os pontos acima serão menos problemáticos quando os computadores quânticos atingirem a tolerância a falhas, mas devem ser considerados no curto prazo. Mesmo sistemas com mapeamentos "mais simples" do que os da química quântica podem enfrentar os mesmos obstáculos, se os Hamiltonianos tiverem muitos termos que não comutam. O método de Krylov é mais útil quando o Hamiltoniano pode ser particionado em relativamente poucos grupos de Pauli que comutam, e quando é fácil de implementar em circuitos de Trotter. Ambas as condições são satisfeitas, por exemplo, para muitos modelos de rede de interesse em física. O KQD é especialmente útil quando muito pouco é conhecido sobre o estado fundamental. Isso decorre de suas garantias inerentes de convergência e de sua aplicabilidade em cenários onde métodos alternativos são inviáveis por falta de conhecimento suficiente sobre o estado fundamental.
Embora o KQD seja uma ferramenta poderosa, os aspectos mais demorados do protocolo — em particular, a estimativa de cada elemento do Hamiltoniano projetado e a sobreposição dos estados de Krylov — representam oportunidades de melhoria. Uma abordagem alternativa envolve combinar os métodos de Krylov com métodos baseados em amostragem, que são o tema da próxima lição.
6. Apêndices
Apêndice I: subespaço de Krylov a partir de evoluções temporais reais
O espaço de Krylov unitário é definido como
para algum passo de tempo que determinaremos adiante. Por ora, suponha que seja par: defina então . Note que, ao projetarmos o Hamiltoniano no espaço de Krylov acima, ele se torna indistinguível do espaço de Krylov
ou seja, aquele em que todas as evoluções temporais foram deslocadas passos de tempo para trás. A razão para essa indistinguibilidade é que os elementos de matriz
são invariantes sob deslocamentos globais do tempo de evolução, uma vez que as evoluções temporais comutam com o Hamiltoniano. Para ímpar, podemos usar a análise para .
Queremos mostrar que, em algum lugar nesse espaço de Krylov, há garantidamente um estado de baixa energia. Fazemos isso por meio do seguinte resultado, derivado do Teorema 3.1 em [3]:
Afirmação 1: existe uma função tal que, para energias no intervalo espectral do Hamiltoniano (isto é, entre a energia do estado fundamental e a energia máxima)...
- para todos os valores de que se encontram afastados de , ou seja, a função é suprimida exponencialmente
- é uma combinação linear de para
Apresentamos uma prova abaixo, que pode ser tranquilamente ignorada a menos que você queira entender o argumento completo e rigoroso. Por ora, focamos nas implicações da afirmação acima. Pela propriedade 3, vemos que o espaço de Krylov deslocado acima contém o estado . Esse é nosso estado de baixa energia. Para entender o porquê, escrevemos na base dos autoestados de energia:
onde é o -ésimo autoestado de energia e é sua amplitude no estado inicial . Expresso em termos disso, é dado por
usando o fato de que podemos substituir por quando ele age sobre o autoestado . O erro de energia desse estado é, portanto,
Para transformar isso em um limitante superior mais fácil de entender, separamos primeiro a soma no numerador em termos com e termos com :
Podemos limitar superiormente o primeiro termo por ,
onde o primeiro passo segue porque para cada na soma, e o segundo passo segue porque a soma no numerador é um subconjunto da soma no denominador. Para o segundo termo, primeiro limitamos inferiormente o denominador por , já que : juntando tudo novamente, obtemos
Para simplificar o que resta, note que para todos esses , pela definição de sabemos que . Limitando superiormente também e , obtemos
Isso vale para qualquer ; portanto, se definirmos como igual ao erro-alvo, o limitante de erro acima converge exponencialmente em direção a esse valor com a dimensão de Krylov . Note também que, se , o termo desaparece completamente do limitante acima.
Para completar o argumento, observamos primeiro que o que foi mostrado acima é apenas o erro de energia do estado específico , e não o erro de energia do estado de menor energia no espaço de Krylov. No entanto, pelo princípio variacional (Rayleigh-Ritz), o erro de energia do estado de menor energia no espaço de Krylov é limitado superiormente pelo erro de energia de qualquer estado no espaço de Krylov; portanto, o resultado acima também é um limitante superior para o erro de energia do estado de menor energia, ou seja, a saída do algoritmo de diagonalização quântica de Krylov.
Uma análise semelhante à acima pode ser realizada levando em conta adicionalmente o ruído e o procedimento de limiarização discutido no notebook. Veja [2] e [4] para essa análise.
Apêndice II: prova da Afirmação 1
O que se segue é em grande parte derivado de [3], Teorema 3.1: sejam e o espaço dos polinômios residuais (polinômios cujo valor em 0 é 1) de grau no máximo . A solução para
é
e o valor mínimo correspondente é
Queremos converter isso em uma função que possa ser expressa naturalmente em termos de exponenciais complexas, pois essas são as evoluções temporais reais que geram o espaço quântico de Krylov. Para isso, é conveniente introduzir a seguinte transformação de energias dentro do intervalo espectral do Hamiltoniano para números no intervalo : defina
onde é um passo de tempo tal que . Note que e que cresce conforme se afasta de .
Agora, usando o polinômio com os parâmetros a, b, d definidos como , e d = int(r/2), definimos a função:
onde é a energia do estado fundamental. Podemos ver, inserindo , que é um polinômio trigonométrico de grau , ou seja, uma combinação linear de para . Além disso, pela definição de acima, temos que e, para qualquer no intervalo espectral tal que , temos
Referências:
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).