Diagonalização quântica baseada em amostragem de um Hamiltoniano de química
Estimativa de uso: menos de um minuto em um processador Heron r2 (NOTA: Isto é apenas uma estimativa. Seu tempo de execução pode variar.)
Resultados de aprendizagem
Após concluir este tutorial, os usuários deverão entender:
- Como usar o addon SQD do Qiskit para aproximar a energia do estado fundamental de um sistema molecular usando bitstrings amostradas de uma unidade de processamento quântico (QPU).
- Como usar o ffsim para construir um circuito local unitary cluster Jastrow (LUCJ) para simulação de química quântica.
Pré-requisitos
Recomendamos que os usuários se familiarizem com os seguintes tópicos antes de realizar este tutorial:
- Química quântica e segunda quantização
- Uso do primitivo Sampler para amostrar circuitos quânticos
Contexto
Neste tutorial, mostramos como pós-processar amostras quânticas ruidosas para aproximar o estado fundamental da molécula de nitrogênio no comprimento de ligação de equilíbrio, usando o addon SQD do Qiskit para implementar o algoritmo de diagonalização quântica baseada em amostragem (SQD). Mais detalhes sobre o software podem ser encontrados na documentação correspondente, incluindo um exemplo simples para começar.
Este tutorial é recomendado para usuários familiarizados com química quântica: especificamente, familiaridade com o cálculo de energias do estado fundamental de uma molécula. Para uma apresentação detalhada do fluxo de trabalho, consulte o curso de algoritmos de diagonalização quântica.
SQD é uma técnica para encontrar autovalores e autovetores de operadores quânticos, como um Hamiltoniano de sistema quântico, usando computação quântica e computação clássica distribuída juntas. A computação clássica distribuída é usada para processar amostras obtidas de um processador quântico e para projetar e diagonalizar um Hamiltoniano alvo em um subespaço que elas abrangem. Um fluxo de trabalho baseado em SQD tem as seguintes etapas:
- Escolher um ansatz de circuito e aplicá-lo em um computador quântico a um estado de referência (neste caso, o estado de Hartree-Fock).
- Amostrar bitstrings do estado quântico resultante.
- Executar o procedimento de recuperação de configuração autoconsistente nas bitstrings para obter a aproximação do estado fundamental.
O SQD é conhecido por funcionar bem quando o autoestado alvo é esparso: a função de onda é suportada em um conjunto de estados de base cujo tamanho não aumenta exponencialmente com o tamanho do problema.
Química quântica
O Hamiltoniano de um sistema molecular pode ser escrito como
onde os e são números complexos chamados integrais moleculares que podem ser calculados a partir da especificação da molécula usando um programa de computador. Neste tutorial, calculamos as integrais usando o pacote de software PySCF.
Para detalhes sobre como o Hamiltoniano molecular é derivado, consulte um livro-texto sobre química quântica (por exemplo, Modern Quantum Chemistry de Szabo e Ostlund). Para uma explicação de alto nível sobre como problemas de química quântica são mapeados em computadores quânticos, confira a palestra Mapping Problems to Qubits da Qiskit Global Summer School 2024.
Ansatz local unitary cluster Jastrow (LUCJ)
O SQD requer um ansatz de circuito quântico para extrair amostras. Neste tutorial, usaremos o ansatz local unitary cluster Jastrow (LUCJ) devido à sua combinação de motivação física e compatibilidade com hardware. Usaremos o ffsim para construir o circuito ansatz.
O ansatz LUCJ se adapta a QPUs com conectividade de qubit restrita. Os orbitais de spin são mapeados para qubits de forma que o ansatz não exija roteamento com gates SWAP. O hardware IBM® tem uma topologia de qubit em rede heavy-hex, caso em que podemos adotar um padrão "zig-zag", representado abaixo. Neste padrão, orbitais com o mesmo spin são mapeados para qubits com uma topologia de linha (círculos vermelhos e azuis), e uma conexão entre orbitais de spin diferente está presente a cada 4º orbital espacial, com a conexão sendo facilitada por um qubit ancilla (círculos roxos).
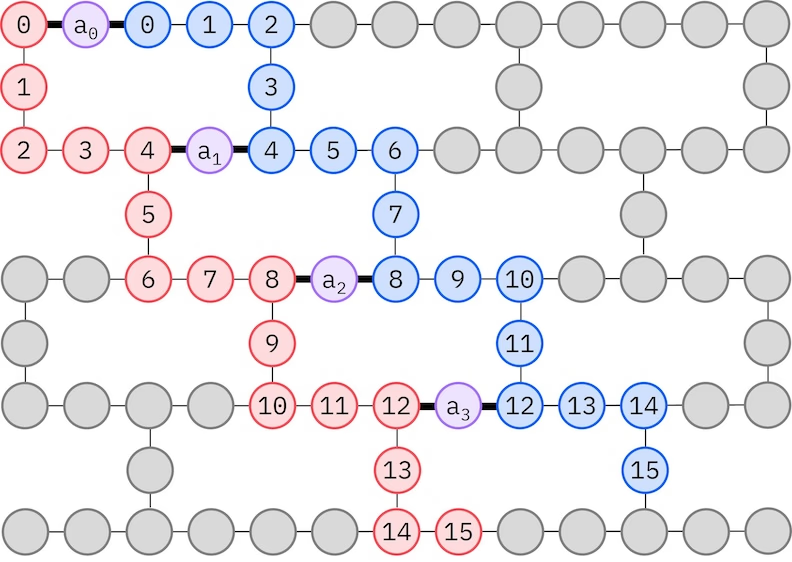
Recuperação de configuração autoconsistente
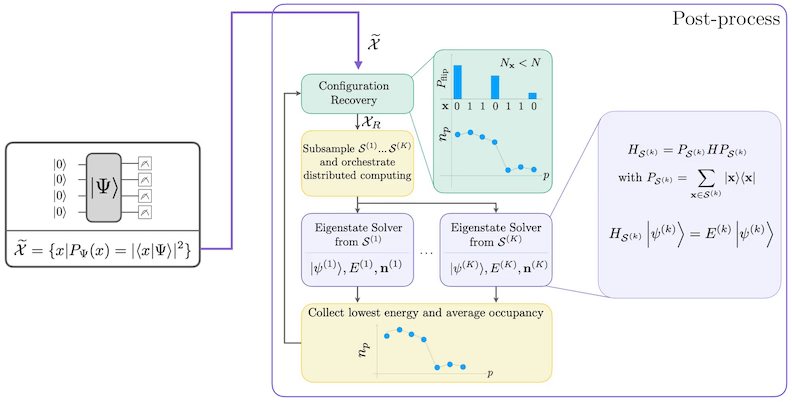
O procedimento de recuperação de configuração autoconsistente é projetado para extrair o máximo de sinal possível de amostras quânticas ruidosas. Como o Hamiltoniano molecular conserva o número de partículas e o spin Z, faz sentido escolher um ansatz de circuito que também conserve essas simetrias. Quando aplicado ao estado de Hartree-Fock, o estado resultante tem um número fixo de partículas e spin Z no cenário sem ruído. Portanto, as metades de spin- e spin- de qualquer bitstring amostrada deste estado devem ter o mesmo peso de Hamming que no estado de Hartree-Fock. Devido à presença de ruído nos processadores quânticos atuais, algumas bitstrings medidas violarão essa propriedade. Uma forma simples de pós-seleção descartaria essas bitstrings, mas isso é desperdiçador porque essas bitstrings ainda podem conter algum sinal. O procedimento de recuperação autoconsistente tenta recuperar parte desse sinal no pós-processamento. O procedimento é iterativo e requer como entrada uma estimativa das ocupações médias de cada orbital no estado fundamental, que é primeiro calculada a partir das amostras brutas. O procedimento é executado em loop, e cada iteração tem as seguintes etapas:
- Para cada bitstring que viola as simetrias especificadas, inverter seus bits com um procedimento probabilístico projetado para aproximar a bitstring da estimativa atual das ocupações orbitais médias, para obter uma nova bitstring.
- Coletar todas as bitstrings antigas e novas que satisfazem as simetrias e subamostrar subconjuntos de um tamanho fixo, escolhido antecipadamente.
- Para cada subconjunto de bitstrings, projetar o Hamiltoniano no subespaço gerado pelos vetores de base correspondentes (veja a seção anterior para uma descrição desses vetores de base), e calcular uma estimativa de estado fundamental do Hamiltoniano projetado em um computador clássico.
- Atualizar a estimativa das ocupações orbitais médias com a estimativa de estado fundamental com a menor energia.
Diagrama de fluxo de trabalho SQD
O fluxo de trabalho SQD é representado no seguinte diagrama:
Requisitos
Antes de iniciar este tutorial, certifique-se de ter o seguinte instalado:
- Qiskit SDK v1.0 ou posterior, com suporte a visualização
- Qiskit Runtime v0.22 ou posterior (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 ou posterior (
pip install qiskit-addon-sqd) - ffsim v0.0.75 ou posterior (
pip install ffsim)
Configuração
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Exemplo em simulador de pequena escala
Neste tutorial, encontraremos uma aproximação do estado fundamental de uma molécula de nitrogênio próxima à sua distância de ligação de equilíbrio. Primeiro usamos um conjunto de base STO-6G pequeno para que possamos simular o experimento e verificar se está funcionando.
Passo 1: Mapear entradas clássicas para um problema quântico
Primeiro, especificamos a molécula e suas propriedades.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Antes de construir o circuito ansatz LUCJ, primeiro realizamos um cálculo CCSD na célula de código a seguir. As amplitudes e deste cálculo serão usadas para inicializar os parâmetros do ansatz.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Agora, usamos ffsim para criar o circuito ansatz. Como nossa molécula tem um estado de Hartree-Fock de camada fechada, usamos a variante com spin balanceado do ansatz UCJ, UCJOpSpinBalanced. Definimos optimize=True no método from_t_amplitudes para habilitar a "compressão" de dupla fatoração das amplitudes (veja The local unitary cluster Jastrow (LUCJ) ansatz na documentação do ffsim para detalhes).
Como o ansatz LUCJ se adapta à conectividade disponível da QPU, precisamos inicializar o backend da QPU antes de criar o ansatz. Por enquanto, criaremos um backend genérico com um mapa de acoplamento heavy-hex e um conjunto de gates ao qual o ansatz LUCJ naturalmente se decompõe. Em seguida, usaremos ffsim.qiskit.generate_lucj_pass_manager para criar um gerenciador de passes especializado para transpilar o ansatz LUCJ para o backend fornecido de acordo com o layout "zig-zag" descrito na seção de contexto sobre o ansatz LUCJ. Esta função utiliza uma heurística de pontuação para minimizar os erros associados ao layout selecionado, o que é importante se o seu backend for uma QPU real ou um simulador com modelo de ruído. Além de retornar o gerenciador de passes, esta função também retorna os pares de acoplamento alfa-beta que podem ser implementados no hardware. Se nem todos os pares puderem ser implementados, ela emite um aviso.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Passo 2: Otimizar para execução em hardware quântico
Em seguida, otimizamos o circuito para um hardware alvo. Normalmente, este passo envolve inicializar o backend de hardware e um gerenciador de passes para esse backend. No entanto, como o ansatz LUCJ é adaptado à conectividade do hardware, já realizamos essas ações no passo anterior. Tudo que resta fazer é executar o gerenciador de passes no circuito para transpilá-lo em um circuito ISA que pode ser diretamente executado na QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Passo 3: Executar usando primitivos do Qiskit
Após otimizar o circuito para execução em hardware, estamos prontos para executá-lo no hardware de destino e coletar amostras para estimativa da energia do estado fundamental. Como temos apenas um circuito, usaremos o modo de execução de Job do Qiskit Runtime e executaremos nosso circuito.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Passo 4: Pós-processar e retornar resultado no formato clássico desejado
Uma métrica útil para avaliar a qualidade da saída da QPU é o número de configurações válidas retornadas. Uma configuração válida tem o número de partículas correto e spin Z correto, o que significa que a metade direita da bitstring tem peso de Hamming igual ao número de elétrons com spin-up, e a metade esquerda tem peso de Hamming igual ao número de elétrons com spin-down. A célula a seguir calcula a fração de configurações amostradas que são válidas.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Todas as bitstrings são válidas porque estamos amostrando o circuito em um simulador sem ruído. Ao executar em uma QPU ruidosa, a fração será menor que um, mas esperamos que seja maior do que a fração que você esperaria se as bitstrings fossem amostradas de forma uniformemente aleatória, calculada na célula a seguir.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Agora, estimamos a energia do estado fundamental do Hamiltoniano usando a função diagonalize_fermionic_hamiltonian. Esta função executa o procedimento de recuperação de configuração auto-consistente para refinar iterativamente as amostras quânticas ruidosas e melhorar a estimativa de energia. Passamos uma função de callback para que possamos salvar os resultados intermediários para análise posterior. Consulte a documentação da API para explicações dos argumentos de diagonalize_fermionic_hamiltonian.
Aqui, usamos o argumento initial_occupancies de diagonalize_fermionic_hamiltonian para especificar a configuração de Hartree-Fock como estimativa inicial das ocupações orbitais no estado fundamental. Essa abordagem é sensata para sistemas onde o estado fundamental tem suporte significativo na configuração de Hartree-Fock, mas pode não ser apropriada em outras situações, embora métodos computacionais mais avançados possam fornecer melhores estimativas iniciais nesses casos. Especificar initial_occupancies também permite que a recuperação de configuração seja executada mesmo que nenhuma configuração válida tenha sido amostrada, como pode ocorrer ao amostrar um circuito grande em uma QPU ruidosa. Sem este argumento, a recuperação de configuração falharia e geraria um erro se nenhuma configuração válida fosse fornecida.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
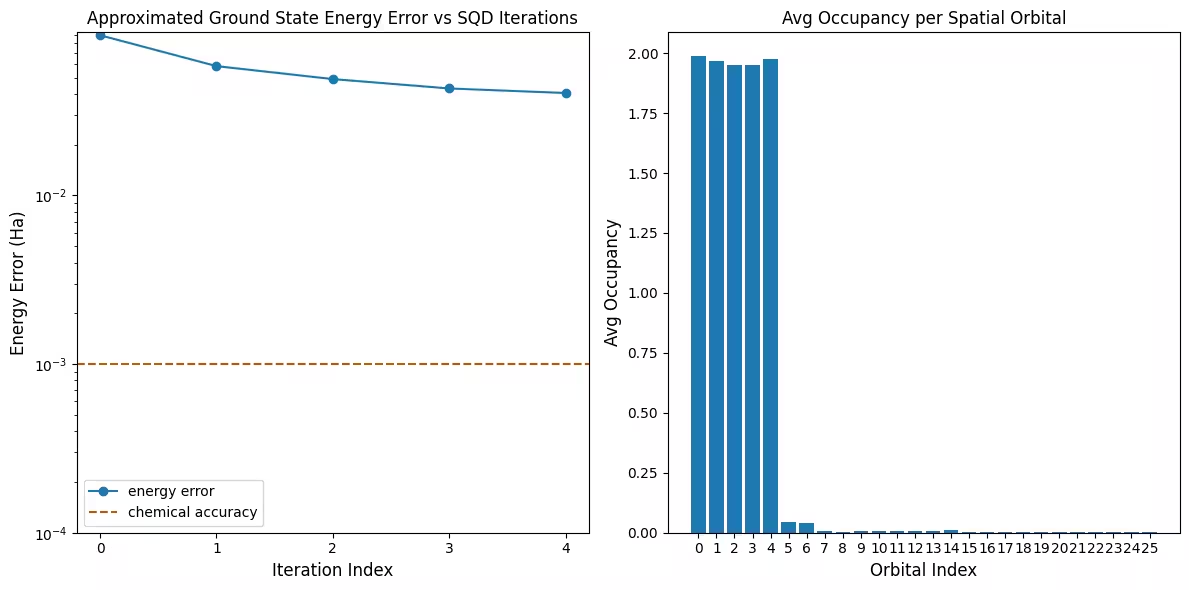
Visualizar os resultados
O primeiro gráfico mostra que nesta simulação já estamos dentro de 1 mH da resposta exata após a primeira iteração (precisão química é tipicamente aceita como sendo 1 kcal/mol 1.6 mH). Este é um sistema pequeno, e como as amostras são sem ruído, a recuperação de configuração não é necessária. Em um sistema maior executado em uma QPU ruidosa, múltiplas iterações de recuperação de configuração podem ser necessárias, e a precisão final pode ser pior. Em geral, a energia pode ser melhorada permitindo mais iterações de recuperação de configuração ou aumentando o número de amostras por lote.
O segundo gráfico mostra a ocupação média de cada orbital espacial após a iteração final. Podemos ver que tanto os elétrons spin-up quanto spin-down ocupam os primeiros cinco orbitais com alta probabilidade em nossas soluções.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Exemplo em hardware real em grande escala
Agora, executamos um exemplo maior em hardware quântico real. Aqui, derivaremos um espaço ativo para a molécula de nitrogênio a partir do conjunto de base cc-pVDZ.
Passos 1-4
Aqui reunimos todos os passos em um único fluxo de trabalho em escala maior, que é então executado em hardware quântico real.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Próximos passos
Se você achou este trabalho interessante, pode se interessar pelo seguinte material:
- Diagonalização quântica de Krylov baseada em amostragem de um modelo de rede fermiônica - um tutorial relacionado usando circuitos de evolução temporal em vez de um ansatz variacional
- Escalar fluxos de trabalho de química SQD com o solver Dice - uma página mostrando como usar o software Dice mais eficiente para diagonalização
- Documentação da API do addon SQD - referência para a função
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - o artigo no qual este tutorial é baseado