Diagonalização quântica de Krylov baseada em amostras de um modelo de rede fermiônica
Estimativa de uso: Nove segundos em um processador Heron r2 (OBSERVAÇÃO: Esta é apenas uma estimativa. Seu tempo de execução pode variar.)
Resultados de aprendizagem
Após concluir este tutorial, os usuários devem compreender:
- Como usar o addon SQD do Qiskit para aproximar a energia do estado fundamental de um modelo de rede usando bitstrings amostradas de uma unidade de processamento quântico (QPU).
- Como usar o ffsim para construir circuitos de evolução temporal para simulação fermiônica.
- Como combinar amostras de múltiplos circuitos para pós-processamento com o algoritmo de diagonalização de Krylov baseada em amostras (SQKD).
Pré-requisitos
Sugerimos que os usuários estejam familiarizados com os seguintes tópicos antes de prosseguir com este tutorial:
- Diagonalização quântica baseada em amostras de um Hamiltoniano de química
- Diagonalização quântica de Krylov de Hamiltonianos de rede
- Primitivas do Qiskit
Contexto
Este tutorial mostra como usar a diagonalização quântica baseada em amostras (SQD) para estimar a energia do estado fundamental de um modelo de rede fermiônica. Especificamente, estudamos o modelo de Anderson de impureza única (SIAM) unidimensional, que é usado para descrever impurezas magnéticas incorporadas em metais.
Este tutorial segue um fluxo de trabalho semelhante ao tutorial relacionado Diagonalização quântica baseada em amostras de um Hamiltoniano de química. No entanto, uma diferença fundamental está em como os circuitos quânticos são construídos. O outro tutorial usa um ansatz variacional heurístico, que é atraente para Hamiltonianos de química com potencialmente milhões de termos de interação. Por outro lado, este tutorial usa circuitos que aproximam a evolução temporal pelo Hamiltoniano. Tais circuitos podem ser profundos, o que torna esta abordagem melhor para aplicações em modelos de rede. Os vetores de estado preparados por esses circuitos formam a base para um subespaço de Krylov, e como resultado, o algoritmo comprovadamente converge de forma eficiente para o estado fundamental, sob suposições adequadas.
A abordagem usada neste tutorial pode ser vista como uma combinação das técnicas usadas em SQD e diagonalização quântica de Krylov (KQD). A abordagem combinada às vezes é chamada de diagonalização quântica de Krylov baseada em amostras (SQKD). Consulte Diagonalização quântica de Krylov de Hamiltonianos de rede para um tutorial sobre o método KQD.
Este tutorial é baseado no trabalho "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", que pode ser consultado para mais detalhes.
Modelo de Anderson de impureza única (SIAM)
O Hamiltoniano SIAM unidimensional é uma soma de três termos:
onde
Aqui, são os operadores fermiônicos de criação/aniquilação para o sítio de banho com spin , são operadores de criação/aniquilação para o modo de impureza, e . , , e são números reais descrevendo as interações de hopping, on-site e hibridização, e é um número real especificando o potencial químico.
Note que o Hamiltoniano é uma instância específica do Hamiltoniano genérico de elétrons em interação,
onde consiste em termos de um corpo, que são quadráticos nos operadores fermiônicos de criação e aniquilação, e consiste em termos de dois corpos, que são quárticos. Para o SIAM,
e contém o restante dos termos no Hamiltoniano. Para representar o Hamiltoniano programaticamente, armazenamos a matriz e o tensor .
Bases de posição e momento
Devido à simetria translacional aproximada em , não esperamos que o estado fundamental seja esparso na base de posição (a base orbital na qual o Hamiltoniano é especificado acima). O desempenho do SQD é garantido apenas se o estado fundamental for esparso, isto é, tem peso significativo em apenas um pequeno número de estados da base computacional. Para melhorar a esparsidade do estado fundamental, realizamos a simulação na base orbital na qual é diagonal. Chamamos essa base de base de momento. Como é um Hamiltoniano fermiônico quadrático, ele pode ser eficientemente diagonalizado por uma rotação orbital.
Evolução temporal aproximada pelo Hamiltoniano
Para aproximar a evolução temporal pelo Hamiltoniano, usamos uma decomposição de Trotter-Suzuki de segunda ordem,
Sob a transformação de Jordan-Wigner, a evolução temporal por equivale a um único portão CPhase entre os orbitais de spin para cima e spin para baixo no sítio de impureza. Como é um Hamiltoniano fermiônico quadrático, a evolução temporal por equivale a uma rotação orbital.
Os estados da base de Krylov , onde é a dimensão do subespaço de Krylov, são formados pela aplicação repetida de um único passo de Trotter, então
No seguinte fluxo de trabalho baseado em SQD, faremos amostragens deste conjunto de circuitos e pós-processaremos o conjunto combinado de bitstrings com SQD. Esta abordagem contrasta com a usada no tutorial relacionado Diagonalização quântica baseada em amostras de um Hamiltoniano de química, onde amostras foram extraídas de um único circuito variacional heurístico.
Requisitos
Antes de iniciar este tutorial, certifique-se de ter o seguinte instalado:
- Qiskit SDK v1.0 ou posterior, com suporte para visualização
- Qiskit Runtime v0.22 ou posterior (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 ou posterior (
pip install qiskit-addon-sqd) - ffsim v0.0.72 ou posterior (
pip install ffsim)
Exemplo em simulador de pequena escala
Passo 1: Mapear o problema para um circuito quântico
Primeiro, geramos o Hamiltoniano SIAM na base de posição. O Hamiltoniano é representado pela matriz e pelo tensor . Então, o rotacionamos para a base de momento. Na base de posição, colocamos a impureza no primeiro sítio. No entanto, quando rotacionamos para a base de momento, movemos a impureza para um sítio central para facilitar interações com outros orbitais.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
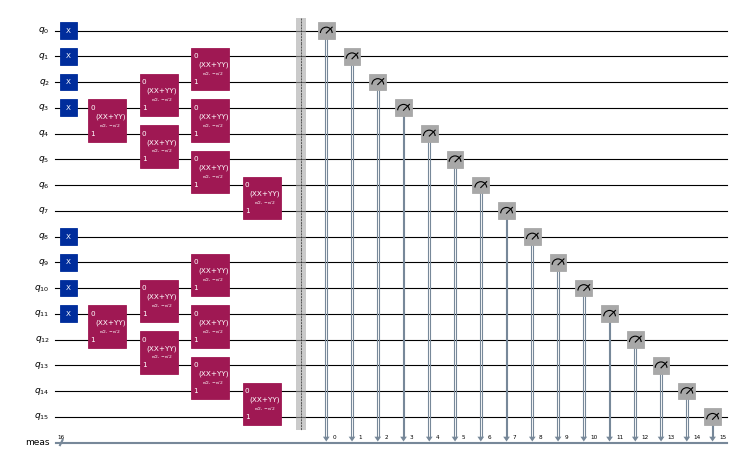
Em seguida, geramos os circuitos para produzir os estados da base de Krylov. Para cada espécie de spin, o estado inicial é dado pela superposição de todas as excitações possíveis dos três elétrons mais próximos do nível de Fermi nos 4 modos vazios mais próximos, partindo do estado , e realizado pela aplicação de sete XXPlusYYGates. Os estados evoluídos no tempo são produzidos por aplicações sucessivas de um passo de Trotter de segunda ordem.
Para uma descrição mais detalhada deste modelo e de como os circuitos são projetados, consulte "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
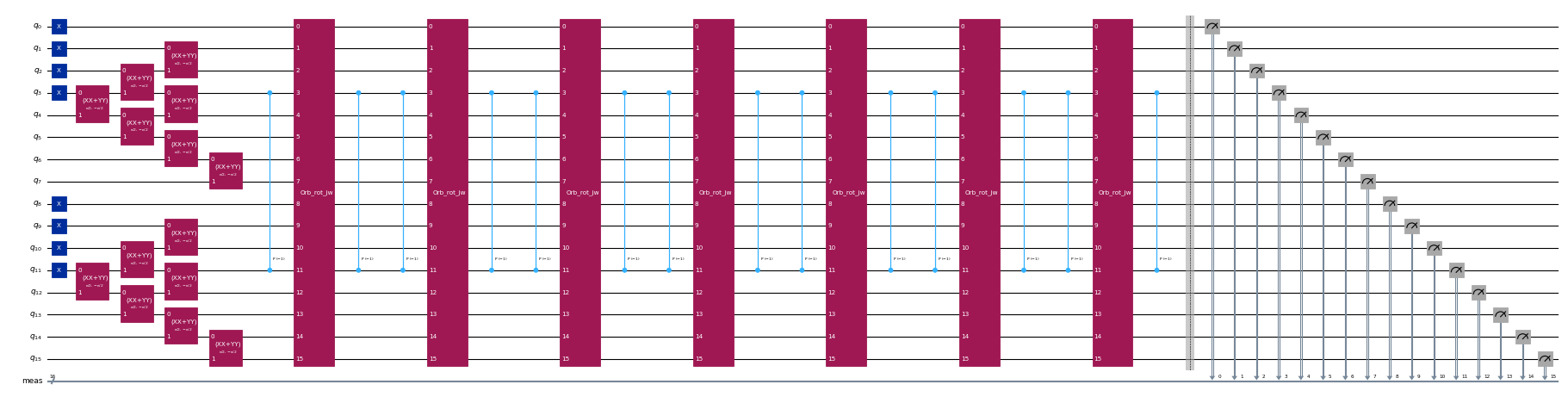
Passo 2: Otimizar o problema para execução quântica
Em seguida, otimizamos o circuito para um hardware alvo. Por enquanto, vamos criar um backend genérico com um número especificado de qubits e um conjunto de portas para o qual os circuitos de evolução temporal se decompõem naturalmente.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Agora, usamos o Qiskit para transpilar os circuitos para o backend alvo.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Passo 3: Executar usando primitivas do Qiskit
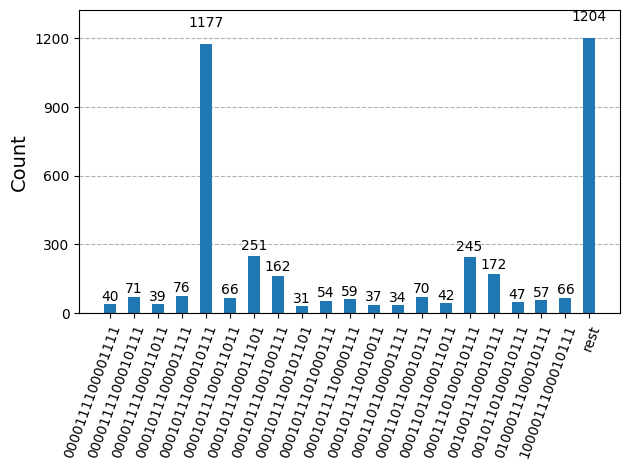
Após otimizar os circuitos para execução em hardware, estamos prontos para executá-los no hardware alvo e coletar amostras para estimativa de energia do estado fundamental. Depois de usar a primitiva Sampler para amostrar bitstrings de cada circuito, combinamos todos os resultados em um único dicionário de contagens e plotamos as 20 bitstrings mais comumente amostradas.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Passo 4: Pós-processar e retornar resultado ao formato clássico desejado
Agora, executamos o algoritmo SQD usando a função diagonalize_fermionic_hamiltonian. Consulte a documentação da API para explicações sobre os argumentos desta função.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
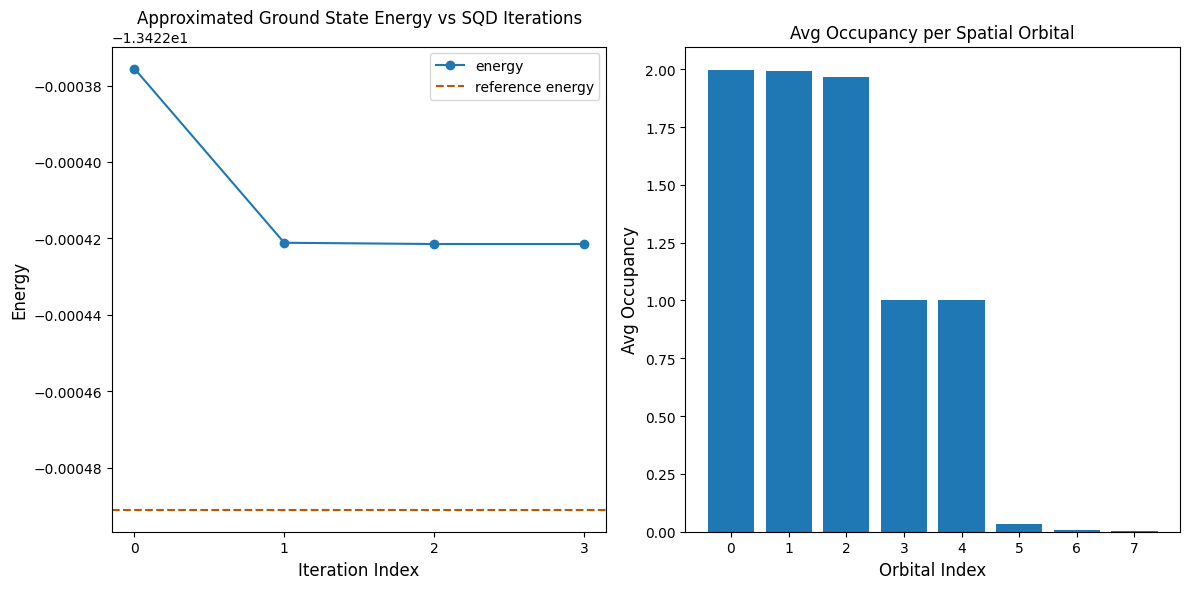
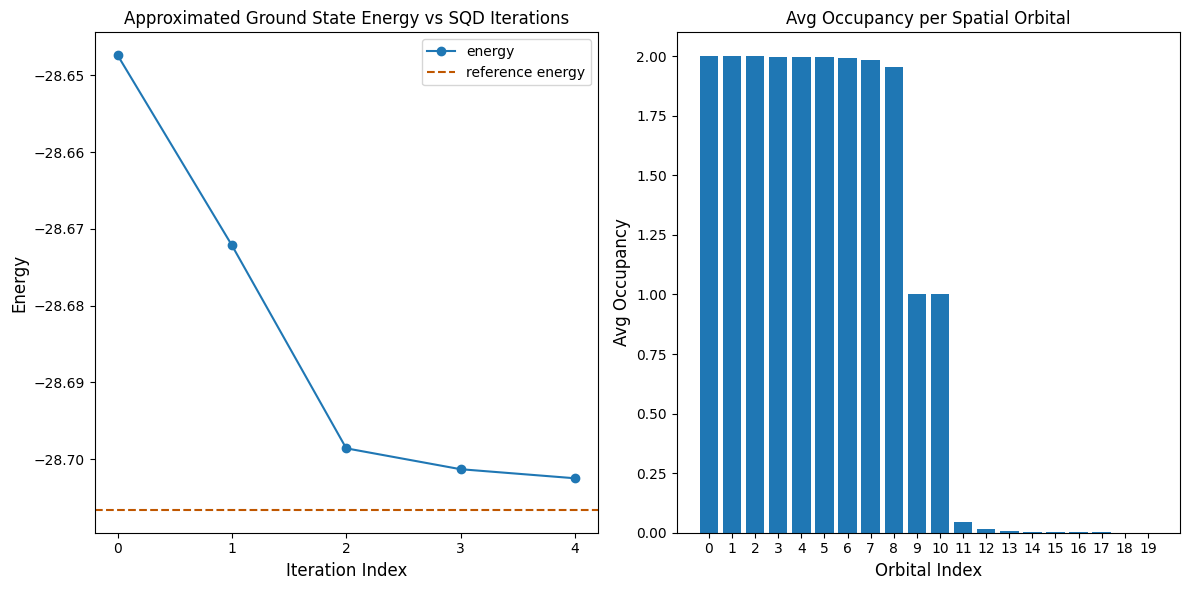
A célula de código a seguir plota os resultados. O primeiro gráfico mostra a energia computada em função do número de iterações de recuperação de configuração, e o segundo gráfico mostra a ocupação média de cada orbital espacial após a iteração final. Como este é um problema de pequena escala, a primeira iteração já nos aproxima muito da energia exata (observe a escala do eixo y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
Verificar a energia
A energia retornada pelo SQD é garantida como um limite superior para a energia verdadeira do estado fundamental. O valor da energia pode ser verificado porque o SQD também retorna os coeficientes do vetor de estado que aproxima o estado fundamental. Você pode computar a energia a partir do vetor de estado usando suas matrizes de densidade reduzida de uma e duas partículas, como demonstrado na célula de código a seguir.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Exemplo em hardware de grande escala
Agora, executamos um exemplo maior em uma QPU real. Para a energia de referência, usamos os resultados de um cálculo DMRG que foi realizado separadamente.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Próximos passos
Se você achou este trabalho interessante, pode se interessar pelo seguinte material:
- Diagonalização quântica baseada em amostras de um Hamiltoniano de química - um tutorial relacionado que usa um ansatz variacional heurístico em vez de circuitos de Trotter
- Diagonalização quântica de Krylov de Hamiltonianos de rede - um tutorial sobre o método KQD
- Documentação da API do addon SQD - referência para a função
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - o artigo no qual este tutorial é baseado