HI-VQE Chemistry - Uma Qiskit Function da Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Consulte a referência da API
As Qiskit Functions são um recurso experimental disponível apenas para usuários dos planos IBM Quantum® Premium Plan, Flex Plan e On-Prem (via IBM Quantum Platform API) Plan. Elas estão em status de versão prévia e sujeitas a alterações.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
Visão geral
Na química quântica, o problema da estrutura eletrônica consiste em encontrar as soluções para a equação de Schrödinger eletrônica — as funções de onda quânticas que descrevem o comportamento dos elétrons do sistema. Essas funções de onda são vetores de amplitudes complexas, onde cada amplitude corresponde à contribuição de uma possível configuração eletrônica.
O estado fundamental é a função de onda de menor energia do sistema e tem uma importância especial no estudo de sistemas moleculares. A abordagem mais precisa para calcular o estado fundamental considera todas as configurações eletrônicas possíveis, mas isso se torna inviável para sistemas maiores, pois o número de configurações cresce exponencialmente com o tamanho do sistema.
O Handover Iterative Variational Quantum Eigensolver (HI-VQE) é um método híbrido quântico-clássico inovador para estimar com precisão o estado fundamental de sistemas moleculares. Ele integra hardware quântico com computação clássica, usando processadores quânticos para explorar eficientemente configurações eletrônicas candidatas e calculando a função de onda resultante em computadores clássicos. Ao gerar funções de onda compactas e quimicamente precisas, o HI-VQE aprimora a pesquisa e a descoberta em química quântica e ciência dos materiais.

O HI-VQE reduz a complexidade computacional do problema da estrutura eletrônica ao estimar o estado fundamental com alta precisão de forma eficiente. Ele se concentra em um subconjunto cuidadosamente selecionado das configurações eletrônicas mais relevantes, otimizando tanto a precisão quanto a eficiência.
Combinando os pontos fortes de computadores clássicos e quânticos, o HI-VQE refina e aprimora iterativamente a estimativa atual da função de onda. Suas técnicas únicas de construção de subespaço ajudam a tornar a seleção de configurações mais eficiente, para que os usuários tenham maior controle computacional e maior precisão nas simulações de química quântica.
Se você quiser conhecer o algoritmo com mais profundidade, pode ler o artigo de pesquisa associado.
Descrição
O número de configurações eletrônicas para um sistema molecular cresce exponencialmente com o tamanho do sistema. No entanto, para certos estados eletrônicos, como o estado fundamental, é comum que apenas uma pequena fração das configurações contribua de forma significativa para a energia do estado. Os métodos de interação de configuração selecionada (SCI) exploram essa esparsidade para reduzir os custos computacionais, identificando e focando nas configurações mais relevantes. Esse subconjunto de configurações é chamado de subespaço.
O HI-VQE aproveita a eficiência inerente dos computadores quânticos para representar sistemas moleculares e auxiliar na busca de subespaços. Ele integra sub-rotinas clássicas e quânticas para resolver o problema da estrutura eletrônica com alta precisão. Ao contrário dos métodos quânticos SCI existentes, o HI-VQE combina treinamento variacional, construção iterativa de subespaço e triagem de configurações por pré-diagonalização para aumentar a eficiência, reduzindo medições quânticas, iterações e custos de diagonalização clássica. O HI-VQE pode, portanto, ser aplicado a sistemas moleculares maiores que requerem mais qubits, e reduz o custo para resolver um problema de determinado tamanho com o mesmo grau de precisão.
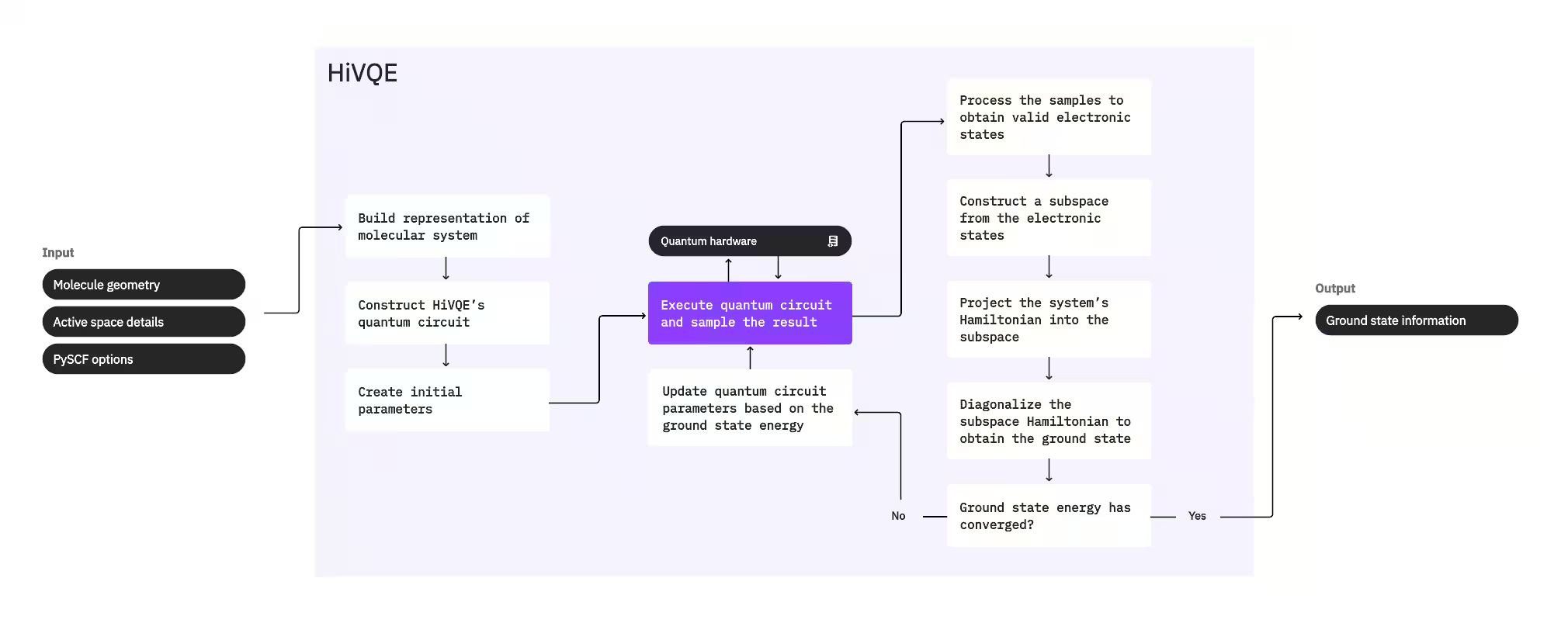
Para calcular o estado fundamental de um sistema, o HI-VQE usa primeiro o pacote de química clássica PySCF para gerar uma representação molecular a partir das entradas fornecidas pelo usuário, como a geometria molecular e outras informações moleculares. Em seguida, entra em um loop de otimização híbrido quântico-clássico, refinando iterativamente um subespaço para representar de forma otimizada o estado fundamental enquanto minimiza o número de configurações incluídas. O loop continua até que critérios de convergência, como tamanho do subespaço ou estabilidade de energia, sejam satisfeitos, após o que a função de onda do estado fundamental calculada e sua energia são produzidas como saída. Esses resultados podem ser usados para construir superfícies de energia potencial precisas e realizar análises mais aprofundadas do sistema.
O loop de otimização se concentra em ajustar os parâmetros de um circuito quântico para gerar um subespaço de alta qualidade. O HI-VQE oferece três opções de circuito quântico: excitation_preserving, efficient_su2 e LUCJ. A otimização é inicializada próximo ao estado de referência de Hartree-Fock devido à sua adequação geral. O circuito é então executado em um dispositivo quântico e as configurações são amostradas do estado quântico resultante antes de serem retornadas como strings binárias. Devido ao ruído do dispositivo quântico, algumas configurações amostradas podem ser fisicamente inválidas, não conservando o número de elétrons ou o spin. O HI-VQE resolve isso usando o processo de recuperação de configuração do pacote qiskit-addon-sqd, para que os usuários possam corrigir configurações inválidas ou descartá-las.
As configurações válidas passam então por uma etapa de triagem opcional para remover aquelas que se prevê contribuir minimamente. Isso reduz a dimensão do subespaço, diminuindo assim o custo da etapa de diagonalização. Se a triagem estiver habilitada, um Hamiltoniano de subespaço preliminar é construído a partir das configurações válidas e uma diagonalização é realizada com critérios de encerramento muito relaxados. Embora a precisão das amplitudes resultantes para cada configuração seja baixa, ela é eficaz para prever quais configurações deixar de fora do subespaço nesta iteração, e é rápida de calcular.
As configurações selecionadas são adicionadas ao subespaço e o Hamiltoniano do sistema é projetado nesse subespaço. O subespaço se atualiza iterativamente, preservando as configurações mais relevantes entre as iterações. Essa abordagem contrasta com métodos alternativos porque o circuito quântico não precisa aproximar o estado fundamental completo a cada etapa.
Em seguida, o Hamiltoniano do subespaço é diagonalizado classicamente para obter o menor autovalor e seu autovetor correspondente, representando uma aproximação do estado fundamental e sua energia. À medida que a qualidade do subespaço melhora com as iterações, o estado fundamental calculado se aproxima melhor do estado fundamental verdadeiro. Uma etapa adicional de triagem pode ser realizada neste ponto para remover quaisquer configurações do subespaço que não tenham uma contribuição substancial para o estado fundamental calculado. Essa etapa garante que o subespaço levado para a próxima iteração seja o mais compacto possível. Isso é avaliado com base nas amplitudes retornadas pela diagonalização, pois elas representam a contribuição de importância de cada configuração para o estado fundamental calculado.
Uma verificação de convergência então determina se um treinamento adicional melhoraria os resultados. Se sim, uma etapa de expansão clássica opcional é realizada, os parâmetros do circuito quântico são atualizados para minimizar ainda mais a energia calculada e o processo se repete. A etapa de expansão clássica gera configurações adicionais para o subespaço, complementando as configurações amostradas do dispositivo quântico. Ela primeiro identifica a configuração com a maior amplitude nos resultados da diagonalização, antes de gerar novas configurações com excitações simples e duplas a partir da configuração identificada. O número desejado dessas configurações é então adicionado ao subespaço.
Uma vez determinado que as iterações convergiram, o HI-VQE retorna o estado fundamental calculado (na forma dos estados no subespaço e suas amplitudes na função de onda do estado fundamental), sua energia e uma medida de variância de energia que indica se o estado calculado forma um autoestado do Hamiltoniano do sistema.
Os usuários podem decidir o circuito quântico utilizado e o número de shots tomados para cada circuito quântico, bem como controlar o tamanho do subespaço ou habilitar a geração clássica de configurações adicionais para auxiliar as configurações geradas pelo quantum. Dessa forma, os usuários podem adaptar o comportamento do HI-VQE para suas aplicações desejadas.
Licenciamento
Observe que o uso desta Qiskit Function é limitado a problemas que requerem no máximo 20 qubits, a menos que seja obtida uma licença que conceda um limite maior.
Envie um e-mail para qiskit.support@qunovacomputing.com se quiser solicitar informações sobre como obter uma licença.
Primeiros passos
Primeiro, solicite acesso à função. Em seguida, autentique-se usando sua chave de API do IBM Quantum® e, assumindo que você já salvou sua conta no seu ambiente local, selecione a Qiskit Function da seguinte forma:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Exemplo
O primeiro exemplo mostra como calcular a energia do estado fundamental de uma molécula de NH3 usando o algoritmo HI-VQE.
Definir a geometria molecular e as opções
A geometria molecular do NH3 é fornecida com coordenadas cartesianas separadas por ";" para cada átomo.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Opções adicionais podem ser definidas e fornecidas para o sistema molecular no seguinte formato de dicionário.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Execute a função com as entradas de geometria e opções.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
É uma boa ideia imprimir o ID do job da Function para que ele possa ser fornecido em solicitações de suporte caso algo dê errado.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Este exemplo usa então 16 qubits com 8 orbitais da base sto3g para uma molécula de NH3. Verifique o status do workload da sua Qiskit Function ou obtenha os resultados da seguinte forma:
print(job.status())
QUEUED
Após a conclusão do job, os resultados podem ser obtidos com a instância result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Para acessar a energia do estado fundamental, use a chave "energy". A chave "eigenvector" fornece os coeficientes CI com a notação de bitstring correspondente da configuração eletrônica armazenada em "states" dos resultados.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Desempenho
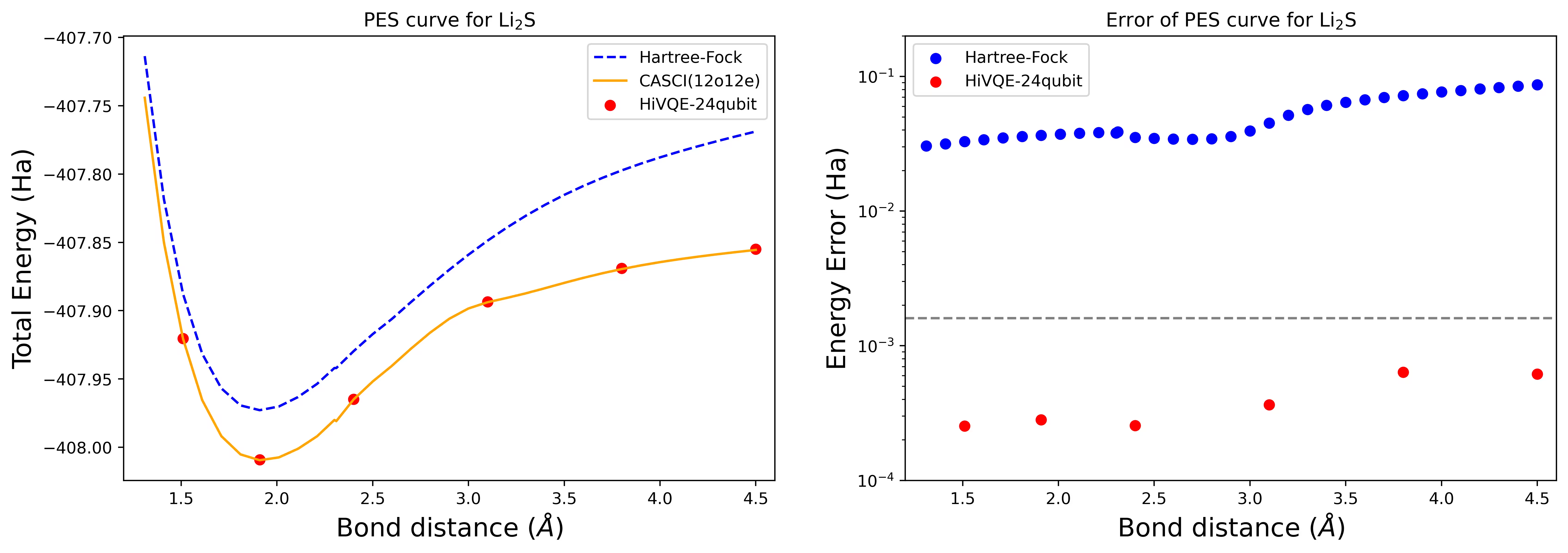
Esta seção mostra os cálculos de benchmark demonstrados do HI-VQE com um caso de 24 qubits para Li2S, um caso de 40 qubits para uma molécula N2 e um caso de 44 qubits para um sistema FeP-NO.
Curva da superfície de energia potencial de dissociação para uma molécula de Li2S com 24 qubits
A curva PES é mostrada com a referência FCI e estimativa inicial de RHF, juntamente com o erro de energia em relação à referência FCI.
 .
.
Os cálculos foram realizados com as seguintes geometrias e opções.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Os pontos vermelhos representam os resultados do cálculo HI-VQE para seis geometrias diferentes, e três geometrias correspondentes a 1,51, 2,40 e 3,80 Ångstroms são fornecidas como entrada na célula acima.
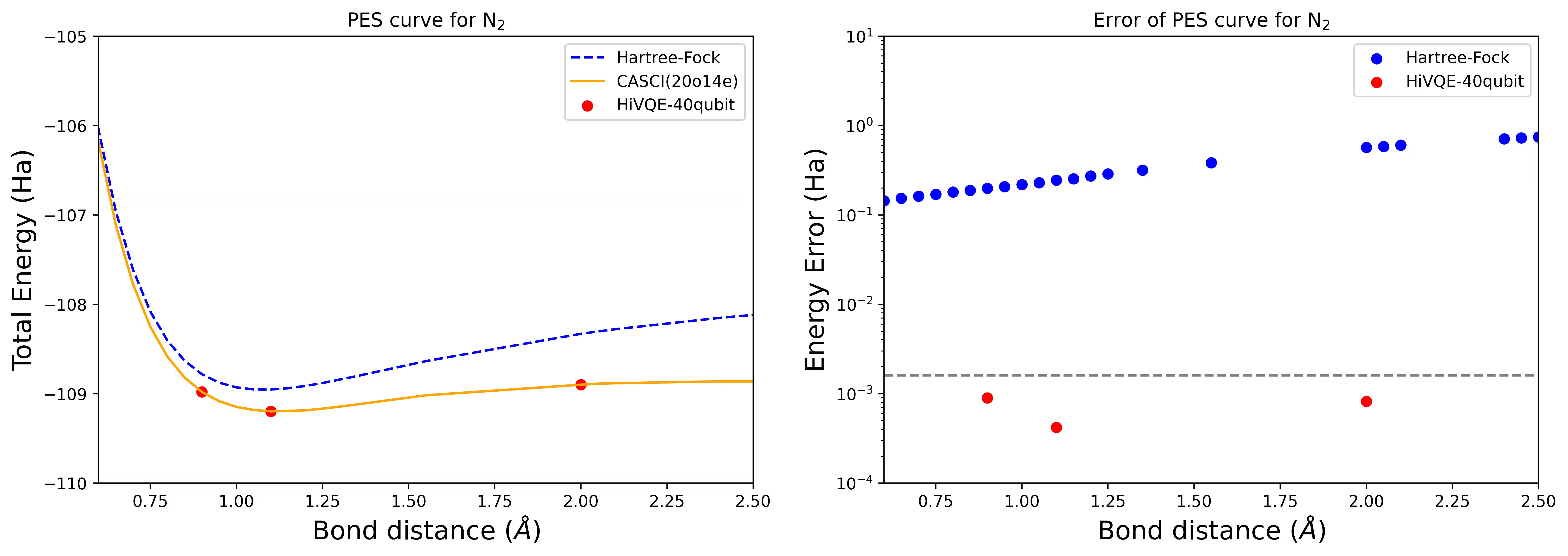
Curva PES de dissociação para uma molécula N2 com 40 qubits
A molécula de nitrogênio foi identificada como um sistema de multireferência com grandes contribuições de energia de correlação além do estado de Hartree-Fock. Realizamos um cálculo de benchmark para a molécula N2 com base cc-pvdz, (20o,14e) usando a seleção de orbital ativo homo-lumo. O número do espaço ativo completo (CAS) para representar este problema é 6.009.350.400. Não é possível obter a solução do problema de autovalor (para energia e estrutura eletrônica) com esse número de estados usando um desktop poderoso (16cpu/64GB). Com o HI-VQE, os usuários podem buscar eficientemente o subespaço de estados CAS para encontrar resultados quimicamente precisos, economizando significativamente recursos computacionais. Os gráficos a seguir mostram a curva PES do cálculo HI-VQE de 40 qubits da dissociação da molécula N2.
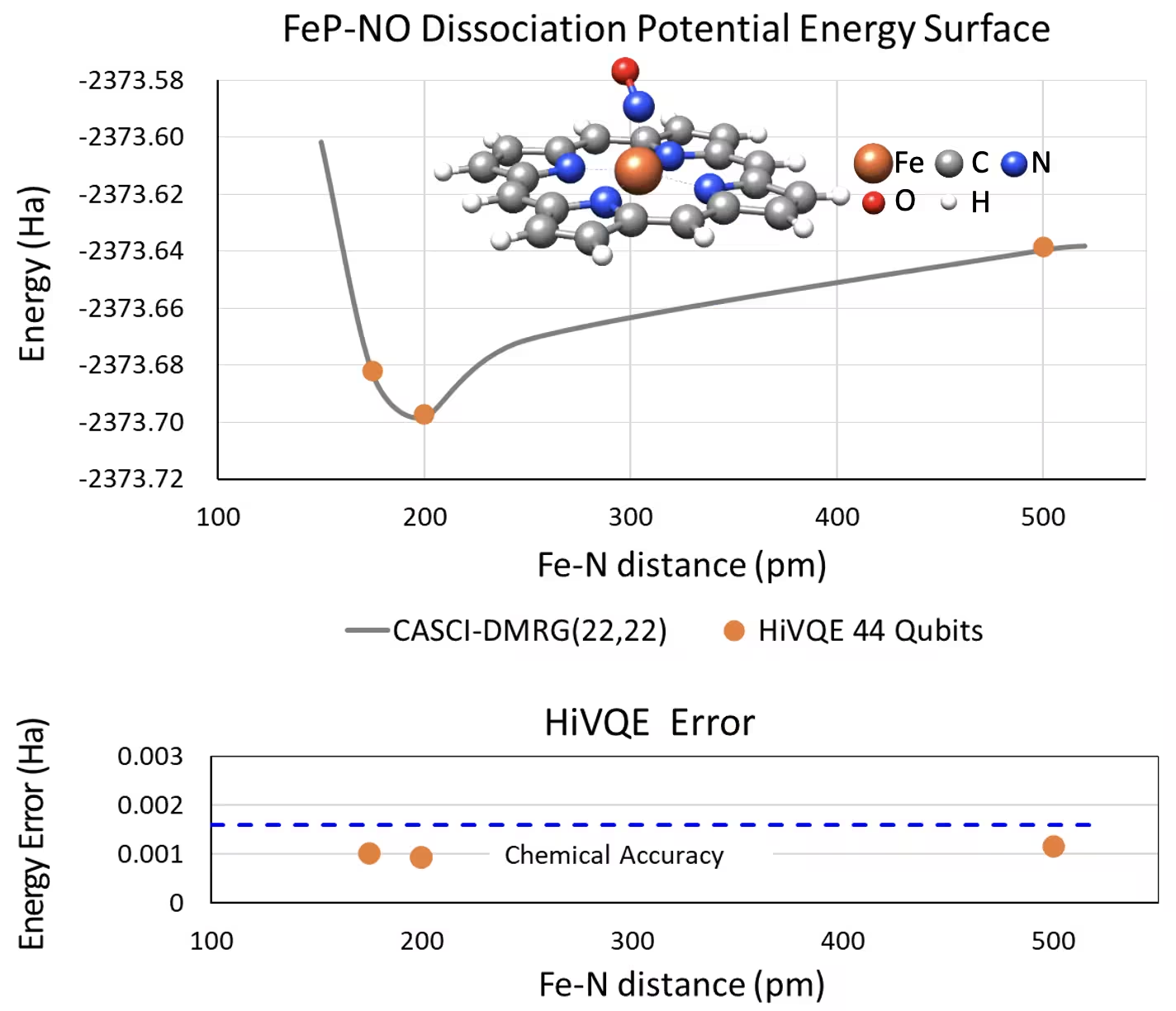
Curva PES de dissociação para o sistema ferro(II)-porfirina pentacoordenado com NO com 44 qubits
Outro sistema químico interessante é um complexo de ferro(II)-porfirina (FeP) com um ligante de óxido nítrico (NO) coordenado, que representa um sistema de metaloporfirina biologicamente relevante que desempenha papéis cruciais em vários processos fisiológicos. Neste exemplo, o HI-VQE foi utilizado para estimar a curva de superfície de energia potencial precisa da interação intermolecular entre FeP e NO (energia do estado fundamental para geometrias com separações diferentes). O sistema combinado tem 450 orbitais e 202 elétrons (450o,202e) com base 6-31g(d) no total. A seleção de orbital ativo homo-lumo foi utilizada para calcular o caso menor a partir do caso real com (22o,22e). Com os seguintes resultados de benchmark, fomos capazes de atingir a precisão química (> 1,6 mHa) com um cálculo de química computacional de referência de última geração em computador clássico, o CASCI(DMRG) (22o,22e).
Benchmarks
- O tamanho exato da matriz é o número de determinantes para a solução exata, como FCI e CASCI.
- O cálculo HI-VQE amostra e calcula o subespaço disso (ou seja, o tamanho da matriz HI-VQE).
- O tempo total inclui o tempo de execução na QPU e as execuções da Qiskit Function com CPU.
- A precisão é estimada a partir da diferença de energia em relação à solução exata.
| Sistema químico | Número de qubits | Tamanho exato da matriz | Tamanho da matriz HI-VQE | E(diff) da exata (mHa) | Número de iterações | Tempo total | Uso do tempo de execução na QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0,08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0,60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0,90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1,10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0,90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1,20 | 21 | 46547 s | 258 s |
Buscar mensagens de erro
Se o seu workload falhar, o status será ERROR e chamar job.result() levantará uma exceção:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Obter suporte
Você pode enviar um e-mail para qiskit.support@qunovacomputing.com para obter ajuda com esta função.
Se você precisar de ajuda com a solução de problemas de um erro específico, forneça o ID do job da Function do job que encontrou o erro.
Próximas etapas
- Solicite acesso à função preenchendo este formulário.
- Visite a referência da API para esta Qiskit Function.
- Experimente o tutorial Compute dissociation PES curve for FeP-NO with HI-VQE.
- Consulte Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Experimente o tutorial Dissociation PES curves with Qunova HiVQE.