Mapeamento
Assista ao vídeo sobre mapeamento por Olivia Lanes, ou abra o vídeo em uma janela separada no YouTube.
Introdução
Nesta aula, vamos focar no primeiro e muitas vezes mais desafiador passo na definição de um programa quântico: mapear um problema para ser executado em um computador quântico. Esse passo abrange como um usuário começa com um problema computacional e o traduz em algo que pode ser resolvido em um computador quântico.
Nas Aulas 2 e 3 deste curso, mencionamos que a etapa de mapeamento é a primeira das quatro etapas totais no framework de padrões Qiskit. Dessas aulas, você pode se lembrar que o objetivo do mapeamento é traduzir ou reescrever um problema computacional em uma função de custo ou valor de expectativa que podemos avaliar usando um computador quântico.
Na Aula 3, discutimos um exemplo concreto com o Max-Cut, um problema computacionalmente difícil, mas muito comum, em otimização combinatória. Nesse exemplo, passamos por várias etapas para traduzir o problema de grafo inicial em algo que pudesse ser resolvido em um computador quântico. Transformamos o problema de encontrar o número máximo de cortes no grafo em uma função de custo, reescrevemos essa função de custo como um Hamiltoniano e, em seguida, preparamos um estado quântico de teste cujo estado fundamental correspondia ao corte máximo. Por fim, construímos um circuito quântico representando o estado quântico de teste de interesse e, em seguida, adicionamos as portas específicas para permitir que o estado evoluísse ao longo do tempo. Essa sequência de passos foi toda parte do mapeamento. Embora os passos exatos fossem exclusivos do problema Max-Cut, o mesmo procedimento geral pode ser aplicado a muitas outras aplicações, como química quântica e simulações quânticas.
O mapeamento pode ser difícil. Não existe uma estratégia única para todos os problemas, então pode ser intimidador. Nesta aula, vamos ver algumas considerações gerais para o mapeamento e, em seguida, mergulhar em alguns problemas exemplares representativos para demonstrar as várias maneiras de mapear um problema para um computador quântico.
Considerações gerais
Embora a estratégia exata usada para mapear um problema a um computador quântico dependa do problema em questão, elas geralmente realizam algumas coisas principais.
Primeiro, você pode precisar simplificar o problema para torná-lo viável. Isso não é exclusivo do quântico — todas as disciplinas científicas usam modelos simplificados para estudar os fenômenos que lhes interessam, ignorando detalhes irrelevantes. Na física, há uma expressão famosa que leva esse princípio ao extremo: "assuma uma vaca esférica." Geralmente é muito difícil descrever um sistema exatamente como ele aparece, mas podemos fazer simplificações razoáveis que ainda podem levar a soluções úteis. Alguns exemplos de como poderíamos fazer isso na computação quântica são limitar o tamanho ou a profundidade do circuito escolhendo um ansatz eficiente para o hardware, truncando evoluções temporais complexas ou negligenciando termos do Hamiltoniano que contribuem pouco para a energia final de um estado quântico.
Segundo, o mapeamento envolve escrever o problema de forma que um computador quântico possa entendê-lo. Isso geralmente envolve responder a estas três perguntas:
- O que nossos qubits vão representar no nosso modelo?
- Nosso problema é contínuo? Como os computadores quânticos são digitais, se o problema for contínuo, precisaremos encontrar uma forma de discretizá-lo.
- Por último, a topologia do problema se alinha com a topologia do hardware? Se não, podemos precisar implementar alguns truques para fazê-lo funcionar.
Vamos abordar a primeira pergunta, que está no cerne de muitas das dificuldades de entender o mapeamento: O que um qubit pode representar?
Qubits podem ser usados para representar muitas coisas. O primeiro, e talvez o mais simples, é um nó em um grafo. Grafos são usados para mostrar conectividade em muitos tipos diferentes de problemas matemáticos, e os nós são um elemento fundamental que representa um ponto ou entidade dentro de uma rede. Dependendo do que a rede inteira representa, isso pode ser uma cidade, uma pessoa ou um ferromagneto em uma rede.
Qubits também podem ser usados para representar bósons e férmions, embora eu precise avisar que um único qubit não é exatamente igual a um bóson ou um férmion — é um pouco mais complicado do que isso, como vamos discutir mais adiante na aula.
Agora estamos chegando a exemplos um pouco mais complicados. Para esses modelos, não faz mais sentido falar em termos de qubits únicos; em vez disso, precisamos de grupos deles para compor algo físico. Por exemplo, um grupo de qubits, aqui representado em uma topologia hexagonal pesada, pode ser usado para representar localizações geométricas de aminoácidos; cadeias de polímeros. Outro exemplo é a simulação de espalhamento de hádrons em modelos de física de altas energias, que pode ser feita simulando a evolução temporal de um pacote de onda hadrônico. Nesse caso, um registrador de qubits pode ser usado para codificar diferentes estados de um campo quântico; o estado de vácuo desse campo, ou um pacote de onda que se propaga sobre esse vácuo.
Mas neste ponto já falamos de forma suficientemente abstrata sobre o desafio à nossa frente. Vamos examinar esses exemplos em detalhe.
Exemplos de mapeamento
Max-Cut
Vamos começar com nosso primeiro exemplo. Um dos problemas de mapeamento mais diretos é o que já cobrimos com alguma profundidade: o exemplo do Max-Cut. Nesse problema, o mapeamento foi bastante fácil para nós porque um qubit era equivalente a um nó no nosso grafo.
Lembre-se que, para mapear o problema Max-Cut, expressamos a função de custo como um Hamiltoniano usando a formulação QUBO. Um Hamiltoniano de função de custo é uma função que codifica a solução ótima do problema no estado fundamental do Hamiltoniano. Para construir o Hamiltoniano de custo, usamos a classe SparsePauliOp no Qiskit para especificar a conectividade do nosso grafo, e a etapa de mapeamento para os operadores quânticos foi feita. E o circuito quântico era simplesmente o ansatz QAOA. Para uma revisão, consulte a aula QAOA em escala de utilidade, onde percorremos tudo isso com muito mais detalhe.
Nessa aula, mesmo no exemplo de escala de utilidade de 100 qubits, a conectividade do grafo já correspondia à topologia do nosso hardware supercondutor. Então não precisamos nos preocupar com como lidar com topologias diferentes. Mas isso nem sempre é o caso. Se tivéssemos um grafo mais complexo ou densamente conectado do que os exemplos que destacamos até agora, precisaríamos implementar uma série de portas SWAP para modificar a conectividade efetiva do hardware. Isso é tratado no segundo passo dos padrões Qiskit, a transpilação, mas deve ser mantido em mente mesmo na etapa de mapeamento.
Dobramento de proteínas
A seguir, vamos explorar um exemplo modelado no artigo chamado "Resource-efficient quantum algorithm for protein folding," escrito pela IBM® e colaboradores da Universidade de New South Wales.
Um pouco de contexto de bioquímica: Proteínas são macromoléculas compostas por longas cadeias de aminoácidos. Essas cadeias se dobram em estruturas complexas que realizam uma grande variedade de funções biológicas. Determinar a estrutura de uma proteína no espaço tridimensional e entender as relações entre estrutura e função estão entre os problemas mais desafiadores da bioquímica hoje. As proteínas se dobram em estruturas úteis devido às interações entre os aminoácidos. À medida que uma estrutura se torce e dobra, aminoácidos que estão distantes uns dos outros ao longo da cadeia podem acabar bem próximos e podem interagir fortemente.
Para modelar isso em um computador quântico, precisamos de um Hamiltoniano descrevendo todas essas interações entre os aminoácidos. Então, podemos prever a estrutura final encontrando o estado que minimizará a energia do nosso Hamiltoniano. Aqui, vamos focar em como cadeias de aminoácidos podem ser modeladas em um computador quântico e como podemos obter distâncias entre aminoácidos para o cálculo de energias de interação. Com isso, teremos reunido todas as contribuições necessárias para o Hamiltoniano que é necessário para simulá-lo em um computador quântico.
Em proteínas reais, os aminoácidos podem ocupar um contínuo de localizações possíveis. No entanto, vamos usar uma simplificação e restringi-los usando um modelo de rede, onde cada aminoácido ocupa um ponto em uma grade. Aqui, os autores usaram uma rede tetraédrica. Nota rápida: aqui estamos codificando a direção das arestas, não os nós em si, como no problema Max-Cut. Cada qubit representa um possível caminho de um único passo ao longo da grade tetraédrica. Note que sítios adjacentes foram rotulados como A ou B por causa de suas diferentes orientações na rede.
A cadeia de proteínas é representada como uma série de curvas ou direções nessa rede. Cada curva entre aminoácidos pode estar em uma das quatro direções, correspondendo às arestas do tetraedro. Essas quatro curvas possíveis são codificadas usando quatro qubits nos estados 0001, 0010, 0100 ou 1000.
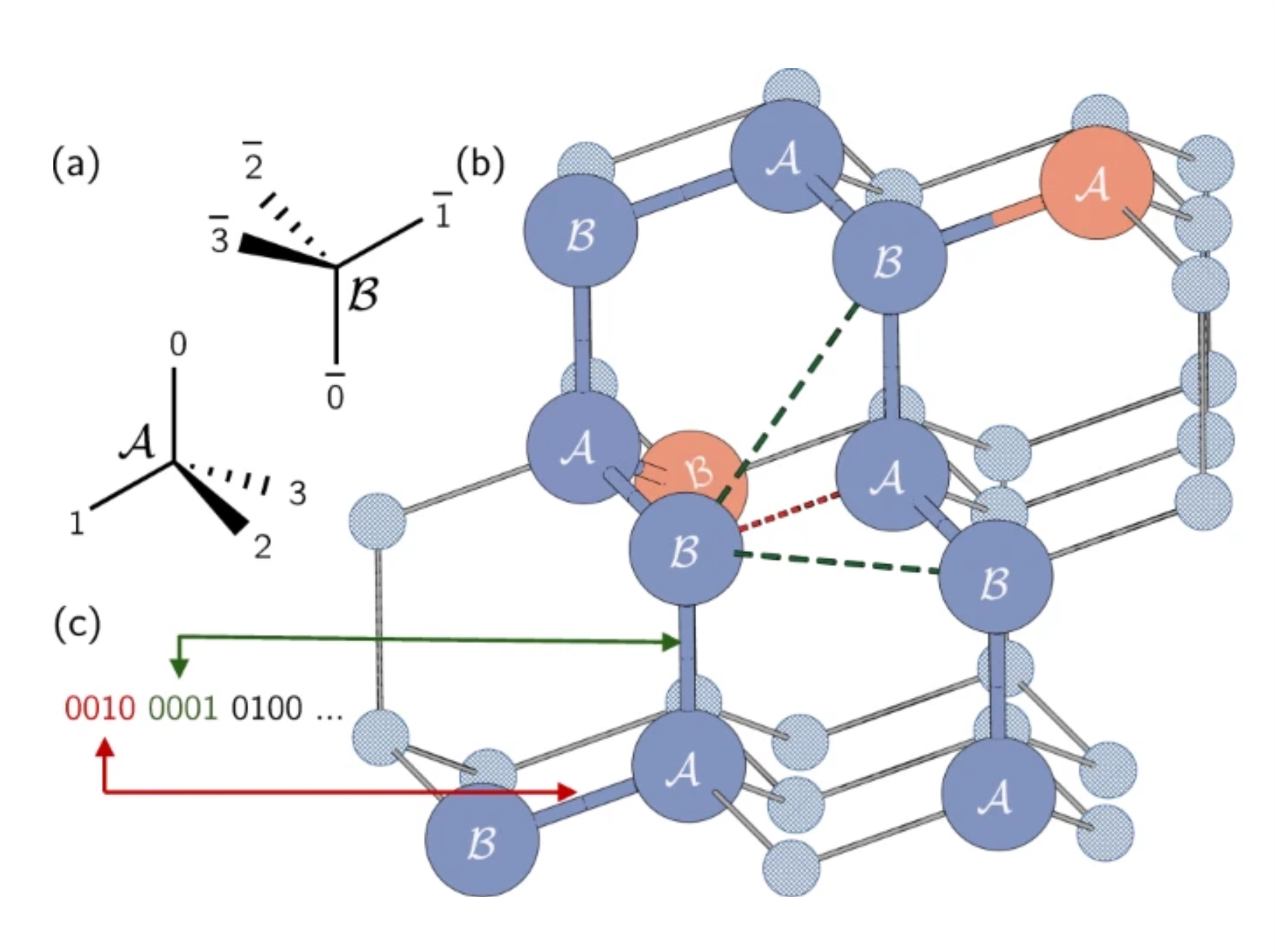
Vamos ver um exemplo na figura acima. Vamos colocar nosso primeiro aminoácido no ponto rotulado "B" circulado em vermelho em nossa rede tetraédrica. A direção para o primeiro aminoácido em relação ao segundo é arbitrária porque o sistema sempre pode ser rotacionado para fazer essa aresta apontar em qualquer direção que quisermos. Então, podemos colocar nosso segundo aminoácido no ponto abaixo do primeiro rotulado "A". Não é tão fácil de ver, mas o caminho do segundo para o terceiro também é arbitrário. As três escolhas resultariam em termos dois seguimentos com um ângulo de aproximadamente 109,5 graus entre eles. Escolher esta segunda aresta simplesmente determina a orientação da nossa proteína no espaço. Portanto, sem perda de generalidade, podemos escolher que as duas primeiras curvas sejam 0001 e 0010.
Com essas simplificações, a configuração da cadeia de aminoácidos é dada por esta expressão:
Até agora, mapeamos as arestas do tetraedro para qubits, e nosso circuito quântico será um ansatz. Agora precisamos considerar como codificar a energia do problema em um Hamiltoniano, de modo que seu estado fundamental nos dê o padrão de dobramento ótimo.
Para qualquer configuração dada, haverá uma energia associada devido às interações entre os aminoácidos. Essas interações são mais fortes quando os dois aminoácidos estão próximos um do outro. Obviamente, os aminoácidos que são adjacentes na cadeia principal da cadeia sempre interagirão uns com os outros. Mas como a proteína pode torcer e dobrar, outros pares de aminoácidos também podem interagir. Os aminoácidos 10 e 20 podem acabar em sítios adjacentes depois que a proteína se dobrou, por exemplo. Portanto, precisamos de uma fórmula para descrever a distância entre os aminoácidos e usando as informações codificadas nos qubits de configuração. Dessa forma, podemos usar a distância de separação para determinar o quão fortemente estão interagindo.
Primeiro, vamos introduzir uma função que indica se uma aresta é ou não usada para a curva no aminoácido . Aqui pode assumir qualquer uma das quatro direções possíveis. Com base na configuração com que começamos acima, podemos escrever essas funções:
Então podemos definir uma diferença no número de curvas rotuladas nas redes A e B do índice ao índice como :
Por que faríamos isso? Bem, acontece que uma curva de do sítio de rede A para B é exatamente cancelada por uma curva de do sítio de rede B para A. Então, para saber a distância do aminoácido no sítio em relação ao do sítio , só precisamos encontrar a diferença entre os passos dados ao longo das arestas dos sítios A e dos sítios B. Como os sítios A e B necessariamente se alternam ao longo da cadeia principal da proteína, isso é capturado pelo . O mesmo argumento se aplica a todos os quatro tipos de aresta. Portanto, a distância total entre os aminoácidos em passos de rede tetraédrica pode ser calculada por esta expressão:
Mas como obtemos o Hamiltoniano dessa longa equação para a distância total entre os aminoácidos? Primeiro, podemos converter da distância em passos de rede para o espaço euclidiano com um pouco de geometria simples:
Em seguida, essas distâncias serão usadas no cálculo da energia da configuração da proteína. Dependendo de nossos objetivos, podemos determinar uma distância de corte abaixo da qual consideramos o par como interagindo, ou podemos fazer algo mais complicado.
Pode não ser óbvio, mas na verdade terminamos a etapa de mapeamento fazendo isso. Os estados dos qubits indicam a "curva" da proteína em cada sítio da rede, e o conjunto de curvas determina a distância entre qualquer par de aminoácidos. Pares de várias espécies de aminoácidos têm interações diferentes, algumas atrativas, outras repulsivas. Se você estiver usando a configuração e as distâncias apenas para determinar se interações conhecidas de aminoácidos estão "ligadas" ou "desligadas", as intensidades dessas interações já foram calculadas e podem simplesmente ser consultadas em uma tabela como esta:
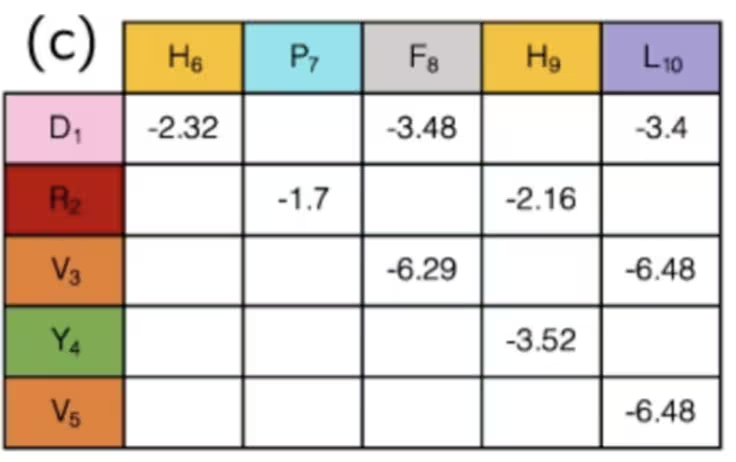
Em resumo, nesse exemplo, os qubits são usados para marcar passos em um caminho ao longo de uma rede, que juntos formam uma cadeia de aminoácidos. Ao simular como eles dobram e se curvam, podemos hopefully encontrar melhores resultados na pesquisa médica. Pulamos como calcular alguns termos desse Hamiltoniano porque eram hiper-específicos para esse problema, enquanto a definição de direções em uma rede pode ser aplicada de forma mais geral. Agora, uma vez que você tenha um Hamiltoniano geral, você vai sempre querer traduzi-lo em operadores de Pauli, que são nativos do computador quântico. É disso que vamos falar a seguir.
Transformação de Jordan-Wigner
Agora vamos explorar como traduzir um sistema de partículas subatômicas em operadores de Pauli.
As partículas subatômicas são divididas em duas categorias — bósons e férmions. Bósons, como fótons ou o Higgs, obedecem a um certo conjunto de regras estatísticas. Férmions, como elétrons ou neutrinos, obedecem a outro. A diferença principal entre eles é que os bósons podem ocupar o mesmo estado — não há limite para quantos bósons podem estar no estado fundamental ou em qualquer estado excitado. Os férmions, por outro lado, são egoístas — eles exigem que cada partícula tenha seu próprio estado quântico.
Os bósons também têm spins inteiros enquanto os férmions têm spin semi-inteiro, como o elétron de spin 1/2, e partículas mais exóticas de spin 3/2. Para descrever um sistema de partículas, precisamos de uma descrição de sua energia. Vamos nos focar nos férmions. Podemos começar com um Hamiltoniano que é escrito em termos do que chamamos de operadores c. Esses são basicamente objetos matemáticos que correspondem a criar ou aniquilar um férmion de um estado no sistema. Eles são frequentemente escritos como e , onde é o operador que cria um férmion no estado e é o operador que destrói um férmion no estado .
Mas lembre-se, os computadores quânticos geralmente operam em uma base de qubits com regras específicas para representar sistemas fermiônicos; eles não trabalham inerentemente na linguagem de operadores fermiônicos. Para preencher essa lacuna, precisamos mapear essa notação fermiônica em operadores de Pauli, que correspondem naturalmente a portas quânticas.
Existem várias dessas transformações para férmions. Uma escolha comum é a transformação de Jordan-Wigner. As codificações de Bravyi-Kitaev e paridade são codificações fermiônicas mais recentes. Operadores bosônicos podem ser transformados usando a transformação de Holstein-Primakoff ou mapeando diretamente os estados de Fock para uma sub-base dos modos bosônicos disponíveis, entre outras opções. Outras codificações também estão sendo ativamente pesquisadas. Por enquanto, vamos focar apenas na transformação de Jordan-Wigner.
A transformação de Jordan-Wigner envolve mapear um único férmion para muitos qubits. Por que não podemos simplesmente atribuir um qubit para representar cada elétron? Isso tem a ver com a indistinguibilidade de férmions idênticos. Qubits são distinguíveis e elétrons não são. Por exemplo, podemos facilmente rotular e identificar qubits individuais em qualquer dispositivo. Mas a indistinguibilidade dos elétrons significa que não podemos rotulá-los de forma alguma. Assim, precisamos realmente rotular os operadores de acordo com os orbitais ocupados, como 1s, 2p, 2p, etc., em vez de elétrons específicos. Portanto, cada qubit desempenha aproximadamente o papel de um orbital na molécula, que está ocupado ou não ocupado. Mas como fazemos isso é um pouco mais complicado. O mapeamento de Jordan-Wigner mantém o controle da antissimetria e garante as estatísticas corretas do sistema fermiônico geral. O mapeamento de Jordan-Wigner expressa os operadores fermiônicos em termos de operadores de Pauli usando estas relações:
O mapeamento de Jordan-Wigner é conceitualmente simples por causa da correspondência um-para-um entre orbitais e qubits. Existem outros mapeamentos que realizam um objetivo semelhante, incluindo o mapeamento de paridade. O cálculo da paridade de um estado requer a consideração de vários qubits. No mapeamento de paridade (e em alguns outros), a interpretação de que um qubit corresponde a um orbital não se mantém. Agora vamos percorrer um exemplo simples. Suponha que queremos calcular a interação de qubit único . Começamos substituindo nossas definições para os operadores de criação e aniquilação.
O comutador . Portanto, a expressão final se torna:
Assim, reescrevemos com sucesso uma expressão fermiônica em termos de operadores de Pauli que o nosso computador quântico poderá entender.
Vamos discutir brevemente como implementaríamos o mapeamento de Jordan-Wigner no Qiskit. É importante entender como esses tipos de transformações funcionam, mas seria impraticável realizá-las manualmente toda vez — especialmente para sistemas de grande porte. Felizmente, o Qiskit facilita isso para nós com a função SparsePauliOp.
Em alto nível, os passos são:
- Use a função
from_listdo SparsePauliOp para criar um operador identidade correspondente ao tamanho do espaço de parâmetros a ser mapeado. - Seguindo a definição dos operadores de criação e destruição mostrados anteriormente, use a função
from_listdo SparsePauliOp para definir operadores de Pauli , , . Isso vai mapear os operadores de criação e aniquilação fermiônicos para operadores de spin de qubit, codificando o número de ocupação fermiônica na base computacional dos qubits. - Gere o Hamiltoniano desejado na base de Pauli aplicando os operadores acima aos orbitais de interesse. Isso geralmente corresponde à criação de uma matriz identidade que representa os orbitais do núcleo (não interagentes) e, em seguida, aplicando os operadores de criação e aniquilação ao espaço ativo, com pesos que correspondem às especificidades do problema em questão.
Agora que entendemos completamente o esquema de mapeamento de Jordan-Wigner, vamos ver um exemplo mais complicado. Você pode se lembrar do artigo intitulado "Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits" da aula anterior. Não vamos percorrer o artigo em detalhe novamente — vamos apenas focar no mapeamento de Jordan-Wigner, que é usado para expressar o Hamiltoniano de sítios de rede com sítios para o modelo de Schwinger na ausência de um campo elétrico.
Aqui, é muito mais difícil identificar especificamente o que um qubit representa neste modelo porque é apenas a coleção de qubits juntos que faz algo físico, neste caso, um pacote de onda. Em vez disso, você pode pensar aproximadamente nos qubits como pedaços discretizados de espaço. Aqui, é o volume da rede no qual cada elemento (célula unitária) compreende dois qubits. Os operadores fermiônicos que vimos no slide anterior descrevem a amplitude da função de onda em um sítio particular. Portanto, nosso Hamiltoniano contém esses operadores de criação e aniquilação fermiônicos. Então, usamos a transformação de Jordan-Wigner para mapear esses operadores para os operadores de Pauli.
onde é o operador de Pauli e é Uma vez que temos um Hamiltoniano escrito nesse formato, a parte difícil da etapa de mapeamento acabou, e agora ele pode ser facilmente escrito em um circuito em operadores de Pauli.
Conclusão
Discutimos quatro exemplos de como técnicas específicas de mapeamento têm sido usadas recentemente no campo da computação quântica, começando do mais simples e avançando até aplicar a transformação de Jordan-Wigner à dinâmica de hádrons. Grande parte desse material foi muito técnico, e se você não viu isso antes, pode parecer muito intimidador. Mas fica mais fácil quanto mais tempo você dedica à prática — por isso este curso se chama Computação Quântica na Prática. Isso não é algo que qualquer um pode simplesmente pegar e rodar desde o início — requer algum estudo, um pouco de coçar a cabeça e, provavelmente, alguns momentos frustrantes. Mas eu te encorajo a ficar com esse desconforto e realmente explorar as questões que surgem à medida que você continua. É a única forma de aprender.