Aprimorando a estimativa de energia de um Hamiltoniano de química com SQD
Neste tutorial, implementamos um padrão Qiskit que mostra como pós-processar amostras quânticas ruidosas para encontrar uma aproximação do estado fundamental de um Hamiltoniano de química: a molécula em equilíbrio no conjunto de bases 6-31G. Seguiremos uma abordagem de diagonalização quântica baseada em amostras para processar amostras tomadas de um ansatz de circuito quântico de 36 qubits (neste caso, um circuito LUCJ). Para considerar o efeito do ruído quântico, é usada a técnica de recuperação de configuração.
O padrão pode ser descrito em quatro etapas:
- Etapa 1: Mapear para o problema quântico
- Gerar um ansatz para estimar o estado fundamental
- Etapa 2: Otimizar o problema
- Transpilar o ansatz para o backend
- Etapa 3: Executar experimentos
- Tirar amostras do ansatz usando a primitiva
Sampler
- Tirar amostras do ansatz usando a primitiva
- Etapa 4: Pós-processar resultados
- Loop de recuperação de configuração autoconsistente
- Pós-processar o conjunto completo de amostras de bitstrings, usando conhecimento prévio do número de partículas e da ocupação orbital média calculada na iteração mais recente.
- Criar probabilisticamente lotes de subamostras a partir das bitstrings recuperadas.
- Projetar e diagonalizar o Hamiltoniano molecular sobre cada subespaço amostrado.
- Salvar a energia mínima do estado fundamental encontrada em todos os lotes e atualizar a ocupação orbital média.
- Loop de recuperação de configuração autoconsistente
Para este exemplo, o Hamiltoniano de elétrons interagentes assume a forma genérica:
/ são os operadores fermiônicos de criação/aniquilação associados ao -ésimo elemento do conjunto de bases e ao spin . e são as integrais eletrônicas de um e dois corpos.
O fluxo de trabalho SQD com recuperação de configuração autoconsistente é representado no diagrama a seguir.
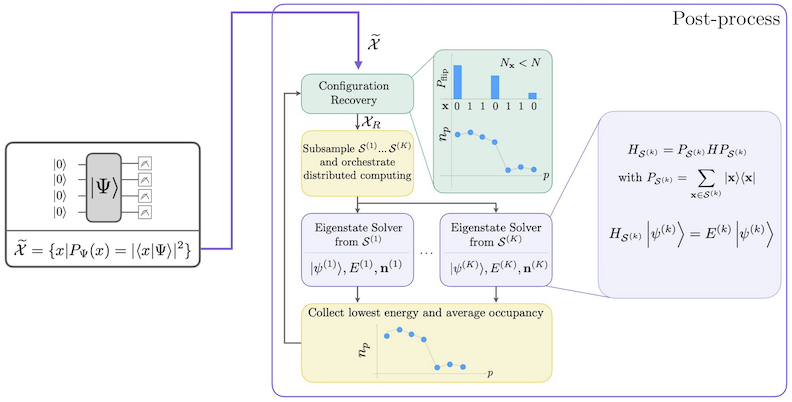
Sabe-se que o SQD funciona bem quando o autoestado alvo é esparso: a função de onda é suportada em um conjunto de estados de base cujo tamanho não aumenta exponencialmente com o tamanho do problema. Neste cenário, a diagonalização do Hamiltoniano projetado no subespaço definido por :
fornece uma boa aproximação do autoestado alvo. O papel do dispositivo quântico é produzir amostras dos membros de apenas. Primeiro, um circuito quântico prepara o estado no dispositivo quântico. A codificação de Jordan-Wigner é usada. Consequentemente, os membros da base computacional representam estados de Fock (configurações eletrônicas/determinantes). O circuito é amostrado na base computacional, produzindo o conjunto de configurações ruidosas . As configurações são representadas por bitstrings. O conjunto é então passado para o bloco de pós-processamento clássico, onde a técnica de recuperação de configuração autoconsistente é usada. No framework SQD, o papel do dispositivo quântico é produzir uma distribuição de probabilidade.
Etapa 1: Mapear o problema para um circuito quântico
Neste tutorial, aproximaremos a energia do estado fundamental de uma molécula . Primeiro, especificaremos a molécula e suas propriedades. Em seguida, criaremos um ansatz local unitary cluster Jastrow (LUCJ) (circuito quântico) para gerar amostras de um computador quântico para estimativa da energia do estado fundamental.
Primeiro, especificaremos a molécula e suas propriedades.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Em seguida, criaremos o ansatz. O ansatz LUCJ é um circuito quântico parametrizado, e o inicializaremos com amplitudes t2 e t1 obtidas de um cálculo CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Usaremos o pacote ffsim para criar e inicializar o ansatz com as amplitudes t2 e t1 calculadas acima. Como nossa molécula tem um estado de Hartree-Fock de camada fechada, usaremos a variante de spin balanceado do ansatz UCJ, UCJOpSpinBalanced.
Como nosso hardware IBM alvo tem uma topologia heavy-hex, adotaremos o padrão zigue-zague para interações de qubits. Neste padrão, os orbitais (representados por qubits) com o mesmo spin são conectados com uma topologia em linha (círculos vermelhos e azuis), onde cada linha assume um formato em zigue-zague devido à conectividade heavy-hex do hardware alvo. Novamente, devido à topologia heavy-hex, os orbitais para spins diferentes têm conexões entre cada 4º orbital (0, 4, 8, etc.) (círculos roxos).
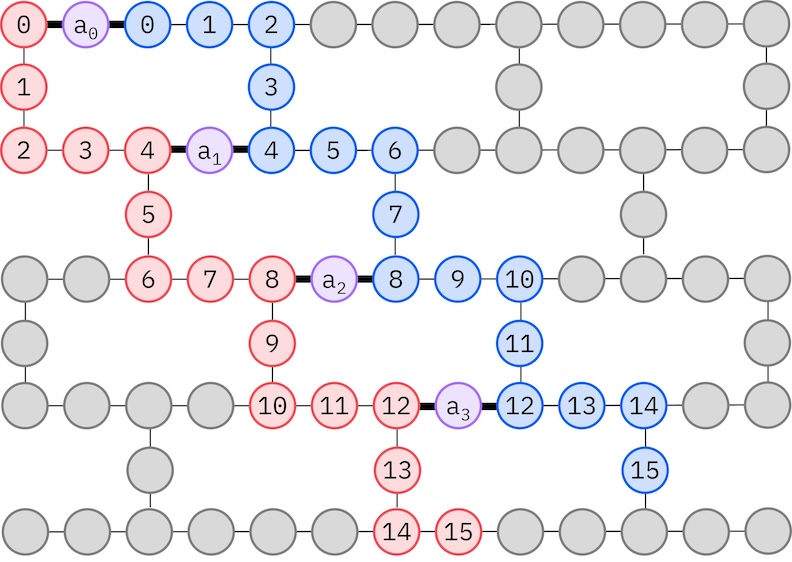
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Etapa 2: Otimizar o problema
Em seguida, otimizaremos nosso circuito para um hardware alvo. Precisamos escolher o dispositivo de hardware a ser usado antes de otimizar nosso circuito. Usaremos um backend falso de 127 qubits do qiskit_ibm_runtime para emular um dispositivo real. Para executar em uma QPU real, basta substituir o backend falso por um backend real. Confira a documentação do Qiskit IBM Runtime para mais informações.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Em seguida, recomendamos as etapas a seguir para otimizar o ansatz e torná-lo compatível com o hardware.
- Selecionar qubits físicos (
initial_layout) do hardware alvo que adere ao padrão zigue-zague descrito acima. Dispor os qubits neste padrão leva a um circuito eficiente compatível com o hardware com menos portas. - Gerar um pass manager em estágios usando a função generate_preset_pass_manager do Qiskit com sua escolha de
backendeinitial_layout. - Definir o estágio
pre_initdo seu pass manager em estágios comoffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITinclui passes do transpilador Qiskit que decompõem portas em rotações orbitais e, em seguida, mesclam as rotações orbitais, resultando em menos portas no circuito final. - Executar o pass manager em seu circuito.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Etapa 3: Executar experimentos
Após otimizar o circuito para a execução em hardware, estamos prontos para executá-lo no hardware alvo e coletar amostras para a estimativa da energia do estado fundamental. Como temos apenas um circuito, usaremos o modo de execução de Job do Qiskit Runtime e executaremos nosso circuito.
Nota: Comentamos o código para executar o circuito em uma QPU e o deixamos para referência do usuário. Em vez de executar em hardware real neste guia, apenas geraremos amostras aleatórias retiradas da distribuição uniforme.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Etapa 4: Pós-processar resultados
Agora, executamos o algoritmo SQD usando a função diagonalize_fermionic_hamiltonian. Veja a documentação da API para explicações sobre os argumentos desta função.
O solver incluído no addon SQD usa a implementação do PySCF de CI selecionado, especificamente pyscf.fci.selected_ci.kernel_fixed_space. O exemplo abaixo também mostra como passar argumentos de palavra-chave para essa função através do solver incluído. Aqui passamos o argumento max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
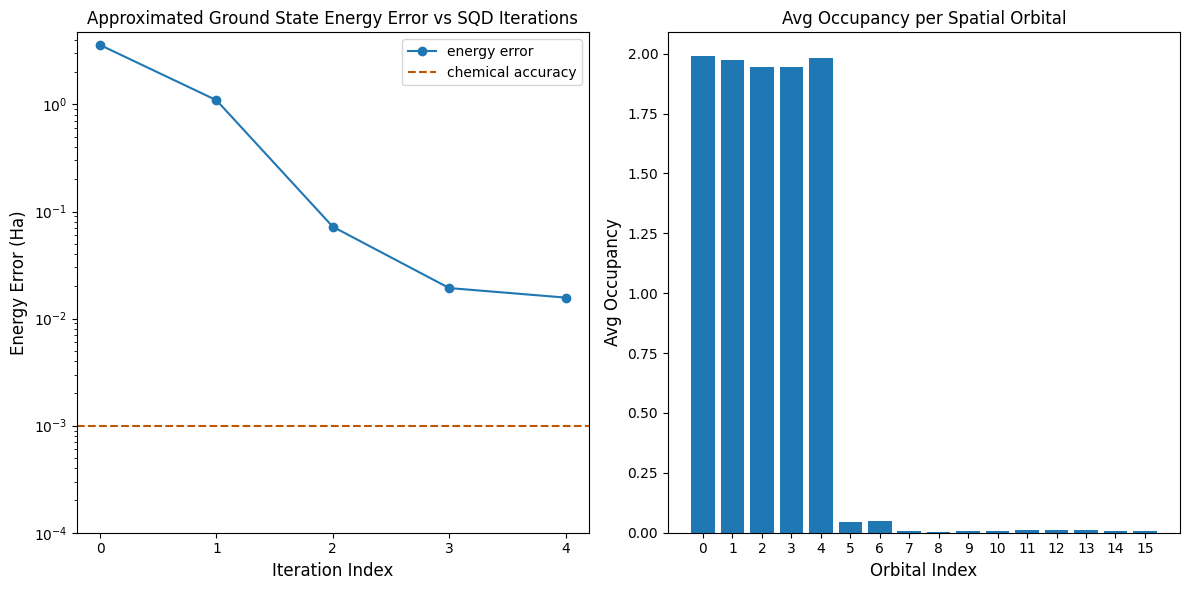
Agora, plotamos os resultados.
O primeiro gráfico mostra que após algumas iterações estimamos a energia do estado fundamental dentro de ~16 mH (a precisão química é tipicamente aceita como sendo 1 kcal/mol 1.6 mH). Lembre-se, as amostras quânticas nesta demonstração eram puro ruído. O sinal aqui vem do conhecimento a priori da estrutura eletrônica e do Hamiltoniano molecular.
O segundo gráfico mostra a ocupação média de cada orbital espacial após a iteração final. Podemos ver que tanto os elétrons spin-up quanto os spin-down ocupam os primeiros cinco orbitais com alta probabilidade em nossas soluções.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha