Algoritmo Variacional Quântico de Autovalores (VQE)
Para este módulo, os alunos devem ter um ambiente Python funcional e as versões mais recentes dos seguintes pacotes instalados:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Para configurar e instalar esses pacotes, consulte o guia Instalar o Qiskit. Para executar tarefas em computadores quânticos reais, os alunos precisarão configurar uma conta IBM Cloud, seguindo os passos no guia Configure sua conta IBM Cloud.
Este módulo foi testado e utilizou aproximadamente 8 minutos de tempo de QPU. Isso é uma estimativa, e o uso real pode variar.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Introdução
Desde o desenvolvimento do modelo quântico-mecânico no início do século XX, os cientistas compreenderam que os elétrons não seguem trajetórias fixas ao redor do núcleo de um átomo, mas existem em regiões de probabilidade chamadas orbitais. Esses orbitais correspondem a níveis de energia específicos e discretos que os elétrons podem ocupar. Os elétrons naturalmente residem nos níveis de energia mais baixos disponíveis, conhecidos como estado fundamental. No entanto, se um elétron absorver energia suficiente, ele pode saltar para um nível de energia mais alto, entrando em um estado excitado. Esse estado excitado é temporário, e o elétron eventualmente retornará a um nível de energia mais baixo, liberando a energia absorvida, frequentemente na forma de luz. Esse processo fundamental de absorção e emissão de energia é importante para entender como os átomos interagem e formam ligações.
Quando os átomos se unem para formar moléculas, seus orbitais atômicos se combinam para formar orbitais moleculares. A disposição e os níveis de energia dos elétrons dentro desses orbitais moleculares ditam as propriedades da molécula resultante e a força das ligações químicas. Por exemplo, na formação de uma molécula de hidrogênio () a partir de dois átomos de hidrogênio individuais, o elétron de cada átomo ocupa orbitais atômicos. À medida que os átomos se aproximam, esses orbitais atômicos se sobrepõem e se combinam para formar novos orbitais moleculares — um com energia mais baixa (um orbital de ligação) e outro com energia mais alta (um orbital antiligante). Os dois elétrons, um de cada átomo de hidrogênio, irão preferencialmente ocupar o orbital de ligação de energia mais baixa, levando à formação de uma ligação covalente estável que mantém a molécula de unida. A diferença de energia entre os átomos separados e a molécula formada, particularmente a energia dos elétrons nos orbitais moleculares, determina a estabilidade e as propriedades da ligação.
Nas seções a seguir, exploraremos esse processo de formação molecular, com foco na molécula . Usaremos um computador quântico real, combinado com técnicas de otimização clássica, para encontrar a energia desse processo simples, porém fundamental. Este experimento fornecerá uma demonstração prática de como a computação quântica pode ser aplicada para resolver problemas em química computacional, fornecendo insights sobre o papel da energia dos elétrons.
VQE - Um algoritmo quântico variacional para problemas de autovalores
Técnicas de aproximação para química - princípio variacional e o conjunto de bases
As contribuições de Erwin Schrödinger para a mecânica quântica não se limitam a introduzir um novo modelo eletrônico; fundamentalmente, ele estabeleceu a mecânica ondulatória ao desenvolver a famosa equação de Schrödinger dependente do tempo:
Aqui, é o operador Hamiltoniano, que representa a energia total do sistema, e é a função de onda que contém todas as informações sobre o estado quântico do sistema. (Nota: é a derivada temporal total, e não incluímos explicitamente o autovalor de energia aqui.)
No entanto, em muitas aplicações práticas — como determinar os níveis de energia permitidos de átomos e moléculas — usamos em vez disso a equação de Schrödinger independente do tempo (equação de autovalor de energia), que é derivada da forma dependente do tempo assumindo um estado estacionário. Um estado estacionário é um estado quântico no qual a densidade de probabilidade de encontrar uma partícula em um determinado ponto do espaço não muda com o tempo.
Nesta forma, representa o autovalor de energia correspondente ao estado quântico . O Hamiltoniano inclui várias contribuições de energia, como a energia cinética de elétrons e núcleos, as forças atrativas entre elétrons e núcleos e as forças repulsivas entre elétrons.
Resolver a equação de autovalor de energia nos permite calcular os níveis de energia quantizados de sistemas atômicos e moleculares. No entanto, para moléculas, resolvê-la exatamente é difícil porque a função de onda , que descreve a distribuição espacial dos elétrons, é complexa e de alta dimensão.
Como resultado, os cientistas usam técnicas de aproximação para obter soluções práticas e precisas. Neste trabalho, focaremos em dois métodos principais:
-
Princípio variacional
Este método aproxima a função de onda e a ajusta para se aproximar o máximo possível da energia alvo, geralmente a energia do estado fundamental do sistema. A ideia central por trás do princípio variacional é simples:
- Se adivinharmos uma função de onda (uma "função tentativa"), a energia calculada a partir dela sempre será igual ou superior à energia do estado fundamental () do sistema.
- Ao ajustar os parâmetros na função tentativa, , podemos obter uma aproximação cada vez melhor da energia do estado fundamental.
- Sua precisão depende muito da escolha da função de onda tentativa . Uma função tentativa mal escolhida pode levar a uma estimativa de energia que está longe de ser precisa.
-
Aproximação do conjunto de bases
O segundo método de aproximação entra no estágio de construção da função de onda — a abordagem do conjunto de bases. Na química quântica, resolver a equação de Schrödinger exatamente para moléculas é quase impossível. Em vez disso, aproximamos a função de onda multi-elétron complexa construindo-a a partir de funções matemáticas mais simples e predefinidas. Um conjunto de bases é essencialmente uma coleção dessas funções matemáticas conhecidas, tipicamente centradas nos átomos da molécula, que são usadas como blocos de construção para representar a forma e o comportamento dos elétrons no sistema. Pense nisso como tentar recriar uma escultura detalhada usando apenas uma coleção de peças LEGO padrão — quanto mais tipos e tamanhos de peças você tiver (quanto maior o conjunto de bases), mais precisamente você pode aproximar a forma original.
Essas funções de base são frequentemente inspiradas pelas soluções analíticas para sistemas simples como o átomo de hidrogênio, tomando formas como funções do tipo Gaussiana ou Slater, embora ainda sejam aproximações. Em vez de trabalhar com os orbitais moleculares completos teoricamente "exatos", mas intratáveis, expressamo-los como uma combinação linear (uma soma com coeficientes) dessas funções de base. Este método é conhecido como a abordagem da Combinação Linear de Orbitais Atômicos (LCAO) quando as funções de base se assemelham a orbitais atômicos. Ao otimizar os coeficientes nesta combinação linear, podemos encontrar a melhor função de onda aproximada e energia possíveis dentro das limitações do conjunto de bases escolhido.
- Quanto mais funções incluídas no conjunto de bases, melhor a aproximação, mas isso tem o custo de maior esforço computacional.
- Um conjunto de bases pequeno fornece uma estimativa aproximada, enquanto um conjunto de bases grande dá resultados mais precisos ao custo de exigir mais recursos computacionais.
Para resumir, para tornar os cálculos viáveis e reduzir o custo computacional, usamos o princípio variacional aproximando a função de onda, o que reduz a complexidade computacional e permite a otimização iterativa para minimizar a energia. Enquanto isso, a abordagem do conjunto de bases simplifica os cálculos representando orbitais atômicos como uma combinação de funções predefinidas, em vez de resolver diretamente para uma função de onda contínua.
Verifique sua compreensão
Considere a função de onda tentativa onde é uma constante de normalização e é um parâmetro ajustável.
(a) Normalize a função de onda tentativa determinando tal que .
Answer
Para normalizar a função de onda tentativa dada:
Use a integral Gaussiana:
defina e obtenha:
(b) Calcule o valor esperado do Hamiltoniano dado por onde , que corresponde a um potencial de oscilador harmônico simples.
Answer
O Hamiltoniano para um oscilador harmônico é:
Valor esperado da energia cinética
Tomando a segunda derivada:
Assim:
Usando resultados padrão da integral Gaussiana:
Valor esperado da energia potencial
Usando:
obtemos:
Valor esperado da energia total
(c) Use o princípio variacional para encontrar o ótimo minimizando .
Answer
Otimize para energia mínima
Diferencie
Resolvendo:
Substituindo em :
o que corresponde à energia exata do estado fundamental do oscilador harmônico quântico.
VQE (Algoritmo Variacional Quântico de Autovalores)
O algoritmo variacional quântico de autovalores (VQE) é o principal método que usaremos para explorar o processo , e aqui veremos o que é o VQE e como ele funciona. Mas vamos primeiro fazer uma pausa e observar uma coisa muito importante por meio da pergunta de verificação.
Verifique sua compreensão
Se já temos tantas estratégias para problemas de química, então por que precisamos de um computador quântico? E qual é o propósito de usar computadores quânticos e clássicos juntos?
Answer
A computação quântica tem a chance de revolucionar a química ao abordar problemas com os quais os computadores clássicos têm dificuldade devido ao escalonamento exponencial dos estados quânticos. Richard Feynman observou famosamente que, para simular a natureza, os cálculos também devem ser quânticos [ref 1].
Por exemplo, simular cafeína com o conjunto de bases mais simples (STO-3G) exigiria bits, muito maior do que o número total de estrelas no universo observável () [ref 2]. Um computador quântico pode descrever os orbitais eletrônicos da cafeína com 160 qubits.
Os computadores quânticos processam naturalmente interações quânticas usando superposição e emaranhamento, que fornecem uma maneira promissora de possibilitar simulações moleculares precisas. Além disso, podemos combinar as vantagens de computadores quânticos (simulação eletrônica) e computadores clássicos (pré/pós-processamento de dados, gerenciamento do processo algorítmico, otimização e assim por diante). Espera-se que esses avanços melhorem a descoberta de materiais, o design de medicamentos e as previsões de reações, reduzindo experimentos caros de tentativa e erro. [ref 3][ref 4]
Se você quiser saber por que computadores quânticos são necessários para problemas de química e por que usar recursos de computação quânticos e clássicos, confira os seguintes artigos:
Agora vamos voltar ao VQE.
O VQE combina o poder dos computadores quânticos com os computadores clássicos, usando fundamentalmente princípios variacionais para obter a energia do estado fundamental do sistema. Para entender o VQE, divida-o primeiro em três partes:
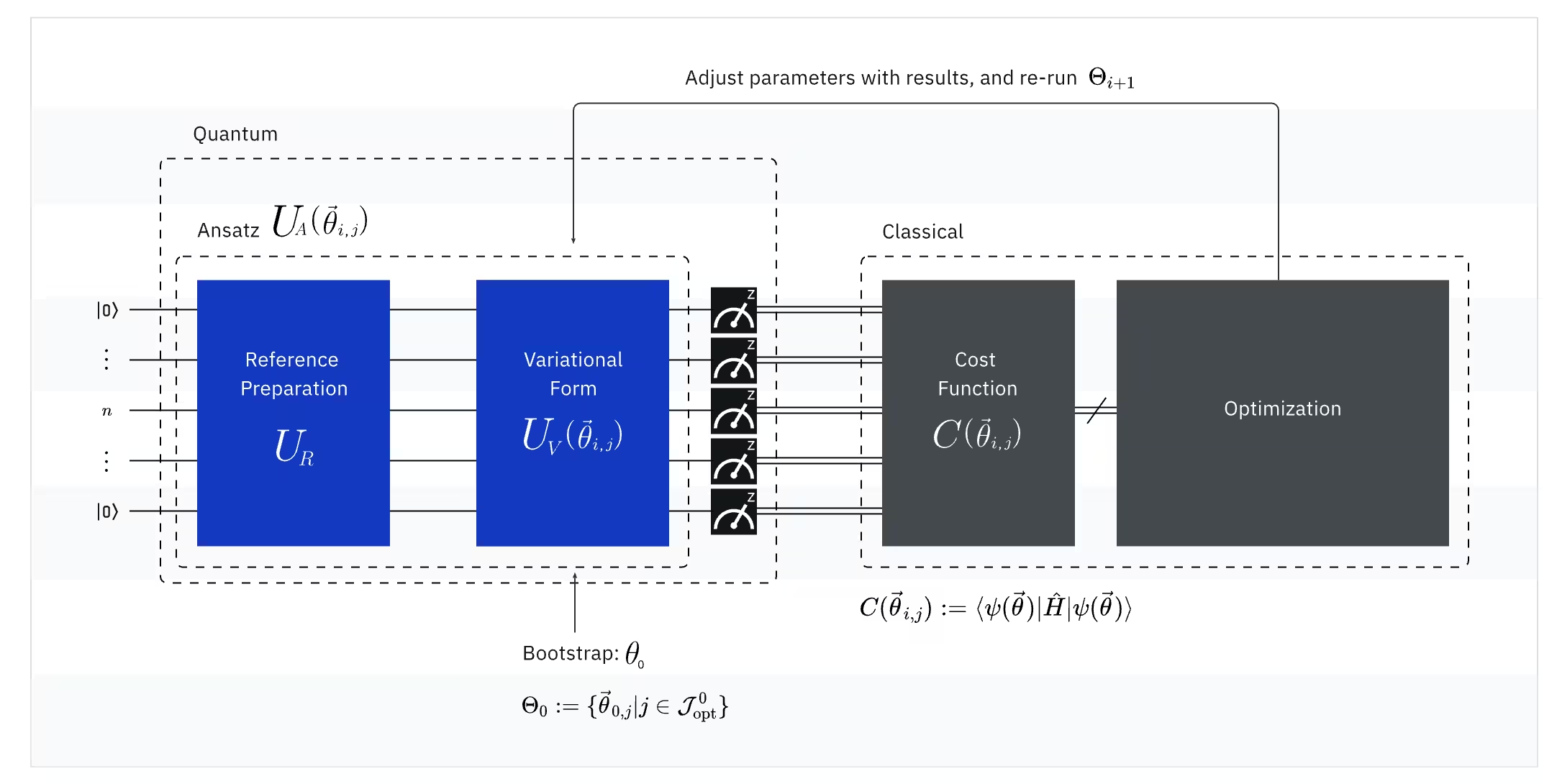
(Quântico) Observável: O Hamiltoniano molecular (energia de uma molécula)
No VQE, o Hamiltoniano molecular/atômico é um observável, o que significa que podemos medir seu valor por meio de um experimento. Nosso objetivo é encontrar a menor energia possível (a energia do estado fundamental) da molécula. Para fazer isso, usamos um estado quântico tentativo, gerado por um circuito quântico parametrizado (ansatz). Medimos o observável e otimizamos o estado quântico até atingirmos a menor energia possível.
O conjunto de bases usado para o Hamiltoniano molecular determina o número de qubits necessários e afeta diretamente a precisão do VQE. Escolher o conjunto de bases correto é fundamental para equilibrar eficiência e precisão. Para simplificar os cálculos sem alterar o conjunto de bases, podemos usar estratégias como impor simetria e redução do espaço ativo. Muitas moléculas têm formas simétricas (como uma borboleta ou floco de neve), o que significa que algumas partes se comportam da mesma forma. Em vez de calcular tudo separadamente, podemos focar apenas nas partes únicas, economizando recursos quânticos, aproveitando assim a simetria. Na redução do espaço ativo, consideramos apenas os orbitais importantes, pois nem todos os elétrons impactam significativamente a energia molecular. Os elétrons próximos ao núcleo permanecem principalmente inalterados, enquanto outros influenciam a ligação. Ao aplicar esses métodos, podemos tornar o VQE mais eficiente enquanto mantemos a precisão.
Uma vez que obtemos um Hamiltoniano molecular usando o conjunto de bases adequado e as estratégias acima, precisamos transformar esse Hamiltoniano em um adequado para computadores quânticos. Mapear problemas para operadores de Pauli pode ser bastante complicado. Isso é especialmente verdadeiro na química quântica, que trabalha com partículas indistinguíveis (elétrons), já que os qubits são distinguíveis. Não entraremos nos detalhes dos mapeamentos aqui, mas remetemos você aos seguintes recursos. Uma discussão geral sobre o mapeamento de um problema para operadores quânticos pode ser encontrada em Computação quântica na prática. Uma discussão mais detalhada sobre o mapeamento de problemas de química em operadores quânticos pode ser encontrada em Química quântica com VQE.
Para este módulo, forneceremos os Hamiltonianos (de um qubit) apropriados para e para que possamos focar no uso do computador quântico. Esses Hamiltonianos de um qubit são preparados usando o conjunto de bases STO-6G e o mapeamento de Jordan-Wigner, que é o mapeamento mais direto com a interpretação física mais simples, pois mapeia a ocupação de um spin-orbital para a ocupação de um qubit. Além disso, usamos uma técnica de redução de qubit usando uma simetria do Hamiltoniano, que usa os padrões de como as ocupações de spin se comportam para reduzir o número de qubits. Para a molécula , assumimos que a distância entre os dois átomos de hidrogênio é 0.735 .
(Quântico) Ansatz: A função de onda tentativa (Como construir um estado quântico trivial com um circuito quântico)
Para o VQE, o ansatz (plural: ansätze) consiste em dois componentes principais. O primeiro é a preparação do estado inicial, que configura o estado do qubit aplicando portas quânticas sem parâmetro variacional. O segundo componente é o circuito quântico parametrizado, um circuito quântico especial com parâmetros ajustáveis, semelhante a botões de rádio. Esses parâmetros serão usados para a última parte — o otimizador clássico — para nos ajudar a alcançar o melhor estado fundamental possível.
Na seção sobre o princípio variacional, aprendemos que a qualidade do estado tentativo afeta a qualidade dos resultados do algoritmo variacional. Isso significa que escolher um bom ansatz é importante no VQE. Mais uma vez, este é um tópico rico e complexo. Não abordaremos os diferentes tipos de ansatz ou suas origens aqui. Se você estiver interessado em aprender mais sobre circuitos quânticos parametrizados e ansatz, pode explorar a lição Ansatz e forma variacional do curso de design de algoritmo variacional, que fornece explicações detalhadas e exemplos de ansätze.
Como vamos usar um Hamiltoniano de um qubit neste módulo, precisamos de um circuito quântico parametrizado de um qubit como ansatz. Veremos três tipos de ansätze de um qubit na seção a seguir. Vamos compará-los e discutir as principais considerações na seleção de um ansatz.
(Clássico) Otimizador: ajuste fino do circuito quântico
Uma vez que o computador quântico mede a energia do observável a partir do ansatz, os parâmetros do ansatz e o valor de energia são enviados ao otimizador clássico para ajuste. Esse processo de otimização é realizado em um computador clássico, tipicamente usando pacotes científicos de uso geral como o SciPy.
O otimizador clássico trata a energia medida como uma função de custo. Em problemas de otimização, uma função de custo (também às vezes chamada de função objetivo) é uma função matemática que mede quão "boa" é uma solução específica. O objetivo do otimizador é encontrar o conjunto de parâmetros que minimiza essa função de custo. No contexto de encontrar a energia do estado fundamental de uma molécula, a própria energia serve como função de custo — queremos encontrar os parâmetros para nosso circuito quântico (nossa "solução") que produzem a menor energia possível. O otimizador clássico usa esse valor de energia medida (o custo) e determina o próximo conjunto de parâmetros otimizados para o ansatz quântico. Esses parâmetros atualizados são então enviados de volta ao circuito quântico, e o processo é repetido. A cada iteração, o otimizador clássico ajusta os parâmetros para tentar reduzir a energia (minimizar a função de custo) até que um critério de convergência predefinido seja atendido, garantindo idealmente que a menor energia possível (correspondente ao estado fundamental da molécula para aquela distância de ligação e conjunto de bases) seja encontrada.
Existem muitas estratégias de otimização fornecidas por pacotes científicos como o SciPy. Você pode encontrar mais na lição Laços de otimização do curso Design de algoritmo variacional. Aqui usaremos COBYLA (Otimização Restrita por Aproximações Lineares), um algoritmo de otimização adequado para paisagens de energia complicadas. Em particular, o COBYLA não tenta calcular um gradiente da função sendo estudada; isso é chamado de otimizador sem gradiente. Imagine que você está tentando encontrar o pico mais alto em uma cordilheira de olhos fechados. Como você não pode ver toda a paisagem, você dá pequenos passos em diferentes direções, verificando se está subindo ou descendo. O COBYLA funciona de maneira semelhante — ele se move pelo espaço de parâmetros, testando diferentes valores, melhorando gradualmente o resultado até encontrar o melhor.
Agora você está pronto para realizar um cálculo VQE. Para isso, tente a pergunta de verificação abaixo, que recapitula o processo geral.
Verifique sua compreensão
Preencha os espaços em branco com os termos corretos para completar o resumo do processo VQE e clique para verificar suas respostas.
O VQE é um algoritmo quântico variacional, que combina o poder da (1) ________ e da computação clássica, usado para encontrar a (2) __________ de uma molécula. O processo começa definindo o (3) __________, que representa a energia total do sistema e atua como o observável nas medições quânticas. Em seguida, preparamos um (4) __________, um circuito quântico com parâmetros ajustáveis que representa a função de onda tentativa da molécula. Esses parâmetros são otimizados usando um (5) __________, um algoritmo clássico que ajusta os parâmetros iterativamente para minimizar a energia medida. Na discussão acima, usamos o otimizador (6) __________, que refina os parâmetros do ansatz sem precisar de cálculos de derivadas. O processo continua até atingirmos a (7) __________, o que significa que encontramos a menor energia possível da molécula.
Banco de palavras:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
Calcule a energia do estado fundamental de um átomo de hidrogênio com VQE
Agora, vamos usar o que aprendemos para calcular a energia do estado fundamental de um átomo de hidrogênio. Ao longo do módulo, usaremos uma estrutura de computação quântica conhecida como "padrões Qiskit", que divide os fluxos de trabalho nas seguintes etapas:
- Etapa 1: Mapear entradas clássicas para um problema quântico
- Etapa 2: Otimizar o problema para execução quântica
- Etapa 3: Executar usando primitivas do Qiskit Runtime
- Etapa 4: Pós-processamento e análise clássica

Geralmente seguiremos essas etapas.
Vamos começar carregando alguns pacotes necessários, incluindo as primitivas do Qiskit Runtime. Também selecionaremos o computador quântico menos ocupado disponível para nós.
Há um código abaixo para salvar suas credenciais no primeiro uso. Certifique-se de excluir essas informações do notebook após salvá-las em seu ambiente, para que suas credenciais não sejam compartilhadas acidentalmente quando você compartilhar o notebook. Consulte Configure sua conta IBM Cloud e Inicialize o serviço em um ambiente não confiável para mais orientações.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
A célula abaixo permitirá que você alterne entre o uso do simulador ou hardware real ao longo do notebook. Recomendamos executá-la agora:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Etapa 1: Mapear o problema para circuitos e operadores quânticos
Iniciamos nosso cálculo VQE definindo o Hamiltoniano para a molécula de hidrogênio () em uma distância de ligação específica. Esse Hamiltoniano representa a energia total do sistema em termos de operadores de qubit, tendo sido produzido e mapeado a partir do sistema molecular usando um procedimento padrão: 1) empregando o conjunto de bases STO-6G (uma coleção específica de funções matemáticas usadas para aproximar os orbitais eletrônicos), 2) aplicando o mapeamento de Jordan-Wigner (uma técnica para traduzir operadores fermiônicos que descrevem elétrons em operadores de qubit), e 3) realizando redução de qubit usando a paridade do Hamiltoniano para simplificar o problema.
Como explicamos anteriormente, as energias do estado fundamental calculadas dependem muito da seleção do conjunto de bases e da geometria molecular (como a distância de ligação). Para esta configuração específica e após essas transformações, o Hamiltoniano de qubit resultante é simples:
Aqui, representa o operador identidade e representa o operador Pauli-Z, atuando em um único qubit. Os coeficientes são derivados das integrais calculadas usando o conjunto de bases STO-6G nesta distância de ligação específica com transformação adequada.
Com este Hamiltoniano definido, agora podemos usar o VQE para calcular sua energia do estado fundamental. É útil comparar nossa energia do estado fundamental calculada com os valores esperados. Para um único átomo de hidrogênio isolado (H), a energia do estado fundamental é exatamente -0,5 Hartree (na ausência de efeitos relativísticos). Vamos calcular a energia exata do estado fundamental do nosso Hamiltoniano de qubit específico conforme definido acima e compará-la com valores conhecidos relevantes.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Em seguida, precisamos de um circuito quântico parametrizado, um ansatz, para preparar uma função de onda tentativa para o estado fundamental. O objetivo é encontrar os parâmetros que minimizam o valor esperado de energia . A escolha do ansatz é crucial porque determina o conjunto de estados quânticos possíveis que nosso circuito pode preparar. Um "bom" ansatz é aquele que é flexível o suficiente para representar um estado muito próximo do verdadeiro estado fundamental do Hamiltoniano que estamos estudando, mas não tão complexo que exija muitos parâmetros ou um circuito muito profundo para os computadores quânticos atuais.
Aqui, tentaremos três ansätze diferentes de um qubit para ver qual deles fornece melhor "cobertura" dos possíveis estados quânticos que um único qubit pode estar. A "cobertura" refere-se à faixa de estados quânticos que o circuito ansatz pode produzir variando seus parâmetros.
Usaremos três ansätze baseados em diferentes combinações de portas de rotação de qubit único:
- Um ansatz com porta de rotação de 1 eixo: Este ansatz usa rotações em torno de um único eixo (). Na esfera de Bloch, isso corresponde a mover-se apenas ao longo de um círculo específico. Este é o menos flexível e cobre um conjunto limitado de estados.
- Dois ansätze com portas de rotação de 2 eixos: Esses ansätze combinam rotações em torno de dois eixos diferentes ( e ). Isso nos permite alcançar uma porção maior da esfera de Bloch, em comparação com uma rotação de eixo único.
Comparando os resultados do VQE obtidos com esses três ansätze, podemos ver como a flexibilidade e a cobertura do espaço de estados do ansatz impactam nossa capacidade de encontrar a verdadeira energia do estado fundamental do nosso Hamiltoniano simplificado. Um ansatz mais flexível tem o potencial de encontrar uma aproximação melhor, mas também pode ser mais difícil para o otimizador clássico.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Agora, vamos gerar 5000 números aleatórios para cada parâmetro e plotar a distribuição de estados quânticos aleatórios, gerados pelos três ansätze com esses parâmetros aleatórios. Você pode pensar nesses parâmetros como rotações em torno de diferentes eixos em uma superfície esférica. Para ver a distribuição do estado quântico, usaremos a esfera de Bloch, uma esfera tridimensional que mostra o estado de um único qubit. Qualquer ponto na esfera representa um possível estado do qubit, onde os polos norte e sul são como os "0" e "1" clássicos, mas o qubit também pode estar em qualquer posição intermediária, mostrando propriedades quânticas especiais como a superposição. Primeiro, prepare as funções necessárias para plotar a esfera de Bloch 3D e prepare 5000 parâmetros aleatórios.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Vamos ver como nosso primeiro ansatz funciona.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
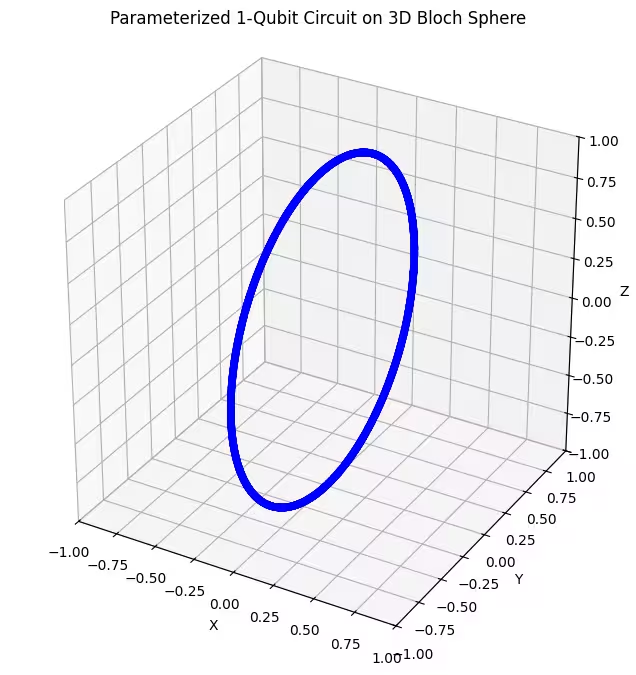
Podemos ver que nosso primeiro ansatz retorna estados quânticos distribuídos em formato de anel na esfera de Bloch. Isso faz sentido, porque fornecemos ao ansatz apenas um parâmetro de rotação único. Portanto, ele pode produzir apenas estados rotacionados em torno de um eixo. Começando do ponto e girando em torno de um eixo, sempre produzirá um anel. Então, vamos verificar nosso segundo ansatz, que tem duas portas de rotação ortogonais - Rx e Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
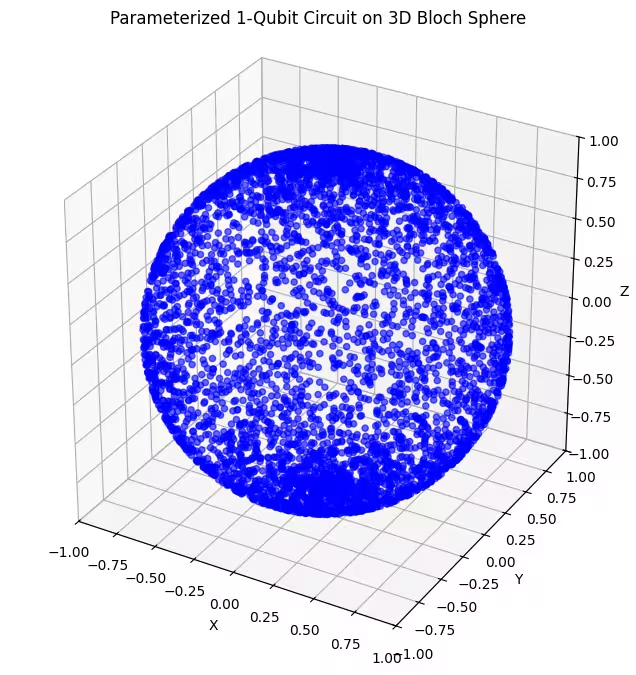
Aqui, podemos ver que nosso segundo ansatz cobre uma porção maior da esfera de Bloch — mas observe que os pontos estão mais concentrados em torno dos polos e mais espalhados em torno do equador. Agora é hora de verificar nosso último ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
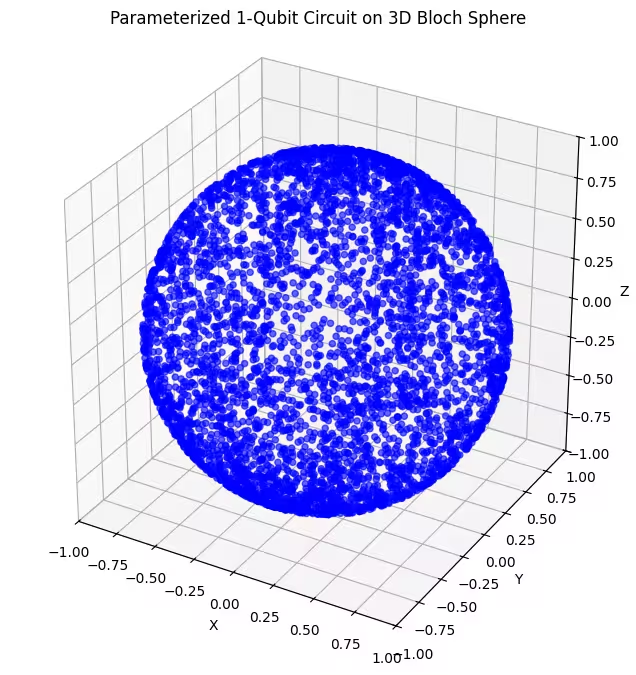
Aqui você pode ver estados quânticos distribuídos de forma mais uniforme gerados pelo nosso último ansatz.
Como mencionado, a melhor coisa a fazer é obter conhecimento sobre o estado fundamental que você está buscando e usar um ansatz bem adequado para sondar estados próximos a esse estado fundamental. Por exemplo, se soubéssemos que nosso estado fundamental estava próximo a um polo, poderíamos selecionar o ansatz 2. Para simplificar, vamos manter o ansatz 3, que sonda uniformemente toda a esfera de Bloch.
Agora que selecionamos nosso ansatz, vamos desenhar o circuito.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Etapa 2: Otimizar para o hardware alvo
Ao executar um cálculo em um computador quântico real, não nos preocupamos apenas com a lógica do circuito quântico. Também nos preocupamos com coisas como quais operações podem ser realizadas por aquele computador quântico específico e onde no computador quântico estão os qubits que estamos usando. Eles estão bem próximos uns dos outros? Estão distantes? Portanto, o próximo passo é reescrever nosso circuito usando portas que são naturais para o computador quântico que usaremos, levando em consideração o layout dos qubits. Isso pode ser feito por transpilação — após esse processo, você pode ver nosso ansatz simples convertido em um conjunto diferente de portas, e nossos qubits abstratos serão mapeados em qubits físicos em um computador quântico real.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Você pode ver que as portas rx, rz do nosso ansatz foram convertidas em uma série de portas rz, sx, que são as portas nativas do nosso backend. Além disso, você pode ver que nosso q0 agora está mapeado no quinto qubit físico. Também precisamos mapear nosso Hamiltoniano de acordo com essas mudanças, como no código a seguir:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Etapa 3: Executar no hardware alvo
Agora é hora de executar nosso VQE em uma QPU real. Para isso, primeiro precisamos de uma função de custo para o processo de otimização, que avalia o valor esperado do Hamiltoniano com um estado quântico, gerado pelo ansatz. Não se preocupe! Você não precisa codificar tudo por conta própria. Preparamos uma função para isso, e tudo que você precisa fazer é executar a célula abaixo.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Por fim, preparamos os parâmetros iniciais para o nosso ansatz e seu processo de otimização. Você pode simplesmente usar todos os zeros ou valores aleatórios. Selecionamos os parâmetros iniciais abaixo, mas sinta-se à vontade para comentar ou descomentar linhas na célula para amostrar parâmetros aleatoriamente, uniformemente de 0 a .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Parabéns! Você acabou de concluir seu primeiro experimento de química quântica com sucesso. Podemos ver uma diferença entre a energia exata do estado fundamental do Hamiltoniano e a nossa, mas como usamos uma técnica padrão de mitigação de erros (que corrige erros de leitura), a diferença é mínima. Este é um ótimo começo!
Nota: Você pode obter um resultado melhor definindo um nível de mitigação de erros usando resilience_level. O valor padrão é 1, e se você definir um valor mais alto, ele usará mais tempo de QPU, mas pode retornar um resultado melhor.
Etapa 4: Pós-processamento
É hora de ver como nosso otimizador clássico funcionou. Execute a célula abaixo e veja o padrão de convergência.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Começamos com um valor inicial bastante bom, de modo que obtivemos um bom valor final em apenas 10 etapas. Você pode ver picos grandes e pequenos, e esta é a característica típica do otimizador COBYLA — ele percorre o espaço como se não pudesse ver a paisagem e ajusta o tamanho dos passos a cada medição.
Verifique sua compreensão
Qual é a sua observação? Qual parte do processo acima está aberta a melhorias para obter resultados mais próximos dos valores teóricos ou mais próximos da energia precisa do estado fundamental do Hamiltoniano? Quais são algumas coisas a considerar para isso?
Answer
A primeira coisa a considerar é a mudança no conjunto de bases usadas no cálculo do Hamiltoniano das moléculas. Como mencionado anteriormente, a energia do estado fundamental do átomo H é -0,5 Hartree, como é bem conhecido, e a base STO-6G que escolhemos não é suficiente para derivar esse valor com precisão.
Escolher um tipo de base mais complexo aumenta o número de qubits usados pelo Hamiltoniano; portanto, precisamos selecionar um ansatz mais complexo e adequado para problemas de química.
O próximo a ser otimizado é o gerenciamento de ruído na QPU. Técnicas de mitigação de erros mais avançadas produzem melhores resultados, mas podem levar mais tempo para serem usadas. Além disso, considere como o shot_number afeta os resultados.
Por fim, melhor desempenho de convergência também pode ser alcançado tentando diferentes otimizadores.
Calcule a energia do estado fundamental da molécula de hidrogênio com VQE
Agora que analisamos o processo geral do VQE usando átomos , calcularemos agora a energia do estado fundamental da molécula de forma mais rápida.
Etapa 1: Mapear o problema para circuitos e operadores quânticos
Aqui também fornecemos um Hamiltoniano de um qubit que usa a base STO-6G e a transformação de Jordan-Wigner, com redução de qubit usando uma simetria do Hamiltoniano. Observe que usamos uma distância atômica entre dois átomos de hidrogênio de 0.735 .
Ao contrário do cálculo de um único átomo de hidrogênio (), para calcular o estado fundamental de uma molécula de hidrogênio (), também devemos considerar a força repulsiva que atua entre os núcleos dos dois átomos de hidrogênio, além da energia associada aos orbitais eletrônicos. Nesta etapa, daremos esse valor como uma constante, e calcularemos esse valor de fato no problema de verificação.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Etapa 2: Otimizar para o hardware alvo
Como o número de qubits usados pelo VQE anterior e pelo Hamiltoniano é o mesmo que o backend a ser usado para execução, usaremos o ansatz existente e sua forma otimizada.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Etapa 3: Executar no hardware alvo
Agora é hora de fazer os cálculos na QPU real. Quase tudo é o mesmo, mas usaremos o ponto inicial apropriado para ajustar ao Hamiltoniano. Além disso, em uma parte iterativa, algumas das configurações do Estimator, que é usado para calcular as expectativas do Hamiltoniano para o ansatz na QPU, serão definidas de forma ligeiramente diferente dos cálculos anteriores. Discutiremos essa mudança mais adiante em uma pergunta de verificação.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Apesar do VQE fornecer teoricamente um limite superior para a verdadeira energia do estado fundamental, implementações práticas em hardware quântico real ou simulado com ruído, bem como aproximações feitas na preparação do Hamiltoniano (como conjuntos de bases ou redução de qubits), podem introduzir erros que às vezes resultam em uma energia medida ligeiramente inferior ao valor teórico exato ou a uma referência numérica específica. Embora haja alguns erros, os resultados parecem ser satisfatórios, especialmente dado o pequeno número de etapas. Agora, vamos concluir este cálculo VQE vendo como o otimizador funcionou.
Etapa 4: Pós-processamento
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Verifique sua compreensão
Vamos calcular a energia de repulsão nuclear da molécula , que incluímos como um valor constante (0,71997 Hartree).

Use a lei de Coulomb e a unidade atômica para garantir que você obtenha o valor em Hartree.
Answer
Como ambos os núcleos de hidrogênio são carregados positivamente, eles se repelem devido à força eletrostática. Essa repulsão é descrita pela lei de Coulomb:
onde é a carga do próton, é a permissividade do vácuo e é a distância entre os dois núcleos, medida em metros ou raios de Bohr em unidades de joules (J).
Para calcular essa energia em Hartrees, precisamos converter a equação acima no sistema de Unidade Atômica (UA). Em UA, , e o raio de Bohr () é 1 e se torna a escala de comprimento fundamental em UA. Com essas simplificações, a lei de Coulomb se reduz a:
onde deve ser medido em raios de Bohr ().
Para converter a separação nuclear dada em para , precisamos desta relação de conversão:
portanto se torna .
Portanto, a energia de repulsão nuclear de um dado é
Calcule a energia de reação de
Agora vamos usar o que obtivemos! Você usou o VQE, um algoritmo variacional quântico de autovalores, para calcular a energia do estado fundamental do átomo e da molécula . O que resta é usar os valores calculados para obter a energia de reação do processo .
A energia de reação é a mudança de energia que ocorre quando substâncias reagem para formar novas substâncias. Imagine que você está construindo algo: às vezes você precisa colocar energia nele (como empilhar blocos), e às vezes a energia é liberada (como uma bola rolando morro abaixo). Na química, as reações absorvem energia (endotérmicas) ou liberam energia (exotérmicas).
A energia de reação do processo pode ser calculada pela seguinte fórmula:
Ao executar a célula abaixo, vamos ver isso visualmente. Aqui usaremos o valor exato do estado fundamental de cada Hamiltoniano, e compararemos a energia de reação da solução exata e dos resultados do VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Como mostrado na figura, embora haja alguns erros, a energia exata do estado fundamental dos Hamiltonianos e a energia de reação calculada usando os resultados do VQE são semelhantes, próximas de -0,2 Hartree.
Deve-se notar aqui que a energia de reação desse processo tem um valor negativo, o que significa que a energia é liberada no processo, e a molécula resultante tem uma energia menor do que dois átomos individuais. 6. Conclusão
Vamos resumir o que aprendemos até agora.
Primeiro, analisamos duas importantes técnicas de aproximação necessárias para resolver problemas de química quântica: o princípio variacional e as escolhas de conjunto de bases, que são ambas fundamentais para o VQE. Exploramos o princípio variacional manualmente, calculando a energia do estado fundamental do oscilador harmônico simples.
Em seguida, exploramos o VQE, um algoritmo amplamente utilizado para calcular a energia do estado fundamental de um sistema quântico. Executamos código para calcular as energias do estado fundamental para o hidrogênio atômico () e a molécula de hidrogênio (). Em particular, aprendemos que é necessário obter o Hamiltoniano molecular adequado para o sistema e transformá-lo em uma forma executável em um computador quântico. Também vimos que o ansatz, um circuito quântico parametrizado, é necessário para preparar estados quânticos de tentativa dentro do VQE, e discutimos a importância de escolher uma estrutura de circuito ansatz adequada. Também aprendemos que o VQE se baseia em um processo de otimização iterativa usando um computador clássico, guiando o circuito quântico para encontrar o estado de menor energia, e vimos como o processo converge.
Por fim, usamos as energias do estado fundamental de e obtidas através do VQE para calcular a energia de reação do processo .
O VQE é um poderoso algoritmo quântico de curto prazo, mas é importante estar ciente de suas limitações. O desempenho do VQE depende muito da escolha do ansatz — encontrar um ansatz eficientemente preparável que possa representar com precisão o verdadeiro estado fundamental torna-se desafiador para moléculas maiores e mais complexas. Além disso, o hardware quântico atual é suscetível a ruídos, o que pode impactar a precisão dos resultados do VQE, particularmente para circuitos mais profundos ou maior número de qubits. Apesar desses desafios, o VQE serve como um algoritmo fundamental, e pesquisas em andamento estão explorando métodos variacionais mais sofisticados e técnicas de mitigação de erros para expandir os limites do que é possível na química quântica em computadores quânticos de curto prazo. Por exemplo, algoritmos como a Diagonalização Quântica Baseada em Amostras (SQD) estão sendo desenvolvidos, que aproveitam amostras obtidas de circuitos quânticos combinadas com diagonalização clássica em um subespaço para melhorar a estimativa de energia e abordar algumas das limitações enfrentadas pelo VQE, particularmente em relação à eficiência de medição e robustez ao ruído.
Revisão e questões
Conceitos críticos:
- O algoritmo quântico variacional é um paradigma de computação no qual um computador clássico e um computador quântico trabalham juntos para resolver um problema.
- No VQE, começamos com um Hamiltoniano do nosso sistema e o mapeamos em qubits para execução no computador quântico. Selecionamos um circuito quântico parametrizado, um ansatz, e fazemos medições repetidas, variando os parâmetros do ansatz, até atingir o menor valor de energia. A busca pelo espaço de parâmetros é feita usando um otimizador clássico. Para obter bons resultados, é necessário selecionar um bom ansatz e um otimizador adequado.
- A energia de reação é a mudança total de energia em uma reação química, determinada pela diferença entre a energia dos reagentes e dos produtos.
Verdadeiro/falso
- O princípio variacional afirma que o valor esperado da energia para qualquer função de onda tentativa é sempre maior ou igual à verdadeira energia do estado fundamental.
- Um conjunto de bases é uma coleção de funções usadas para aproximar funções de onda quânticas.
- O VQE é um algoritmo quântico usado para resolver exatamente a equação de Schrödinger para um dado Hamiltoniano.
- No VQE, um circuito quântico parametrizado (um ansatz) é usado para preparar funções de onda tentativas.
- A escolha do otimizador no VQE (por exemplo, COBYLA, SPSA ou ADAM) não impacta a qualidade do resultado.
- O
Estimatordo Qiskit é usado para calcular diretamente os valores esperados dos Hamiltonianos no VQE.
Questões de múltipla escolha:
- Qual é o propósito do Hamiltoniano no VQE?
- A) Para gerar estados quânticos aleatórios
- B) Para determinar a energia dos estados quânticos
- C) Para otimizar circuitos quânticos
- D) Para criar emaranhamento
- Qual é o objetivo principal do algoritmo VQE?
- A) Para encontrar a energia do estado fundamental de um Hamiltoniano
- B) Para criar emaranhamento entre qubits
- C) Para realizar a busca de Grover
- D) Para quebrar a criptografia RSA
- Quantos estados quânticos são gerados neste notebook para comparar o ansatz?
- A) 100
- B) 1000
- C) 5000
- D) 10.000
- Por que um otimizador clássico é necessário no VQE?
- A) Para realizar medições quânticas
- B) Para atualizar os parâmetros do ansatz para minimizar a energia
- C) Para emaranhamento de qubits
- D) Para gerar aleatoriedade quântica
- Por que o ansatz é projetado para ser parametrizado?
- A) Para permitir a preparação de estados quânticos
- B) Para permitir que um amplo espaço de estados quânticos seja pesquisado
- C) Para reduzir a complexidade do circuito
- D) Para medir autovalores diretamente
- Qual das seguintes é a afirmação mais correta sobre como escolher um bom ansatz?
- A) Um ansatz deve produzir estados distribuídos uniformemente sobre a esfera de Bloch, ou falhará.
- B) Um ansatz deve ser adaptado ao seu sistema para garantir que possa gerar estados próximos ao estado fundamental.
- C) Um ansatz deve produzir estados aleatórios usando seus parâmetros variacionais.
- D) Um ansatz melhor sempre tem mais parâmetros variacionais.
(Opcional) Apêndice: Sobrecarga do otimizador por complexidade do ansatz
O VQE enfrenta vários desafios bem conhecidos [ref 6], e os seguintes estão relacionados ao que aprendemos acima.
- Desafios na seleção do ansatz
Existe um desafio inerente na seleção do ansatz variacional correto. Ansätze inspirados na química (como UCCSD) fornecem precisão física, mas requerem circuitos profundos, enquanto ansätze eficientes em hardware têm circuitos mais rasos, mas podem carecer de interpretabilidade física. Além disso, muitos ansätze introduzem parâmetros variacionais excessivos que contribuem pouco para melhorar a precisão, mas aumentam significativamente a dificuldade de otimização.
- Dificuldades de otimização
A paisagem de otimização do VQE pode ter regiões onde os gradientes desaparecem exponencialmente (platôs estéreis), tornando difícil para os otimizadores clássicos atualizar os parâmetros variacionais eficientemente. Para isso, os pesquisadores tentaram usar diferentes tipos de otimizadores — baseados em gradiente e sem gradiente, mas ambos enfrentam desafios. Os otimizadores baseados em gradiente sofrem de platôs estéreis, enquanto os métodos sem gradiente requerem um grande número de avaliações de função.
- Sobrecarga do otimizador
Mais um desafio bem conhecido é a sobrecarga do otimizador, que está relacionada à escala do problema. Os circuitos quânticos necessários para o VQE crescem em profundidade e complexidade conforme o tamanho do problema aumenta; isso normalmente também aumenta o número de parâmetros a otimizar. O processo de otimização torna-se intratável conforme o número de parâmetros aumenta, levando à convergência lenta e dificuldades em encontrar a solução ótima.
Aqui, veremos esses desafios usando o VQE para uma molécula , com dois tipos diferentes de ansätze.
(Nota: Isso pode levar mais tempo de QPU, então sinta-se à vontade para usar um simulador para isso se não tiver tempo suficiente.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Agora vamos executar um VQE com um ponto inicial composto de uns, com um máximo de 20 etapas, e comparar a convergência de ambas as execuções.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

O gráfico acima demonstra claramente que o processo de otimização do ansatz com mais variáveis leva mais tempo para alcançar uma convergência estável.
Em vez de depender de circuitos simples de qubit único e um ansatz direto, a complexidade da otimização aumenta quando circuitos quânticos maiores e ansätze com estrutura mais complexa são necessários. Isso destaca um desafio bem conhecido nos VQEs: a sobrecarga do otimizador.
Os pesquisadores continuam a desenvolver várias metodologias avançadas que podem usar computadores quânticos para problemas de química. Você pode acessar uma variedade de materiais educacionais em IBM Quantum Learning.
Referências
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)